- Record: found
- Abstract: found
- Article: found
Syndromes microdélétionnels (syndrome de Williams et syndrome de la délétion 22q11) au CHU Hassan II de Fès: à propos de 3 observations Translated title: The Cri du Chat syndrome: report of an observation
Read this article at
Abstract
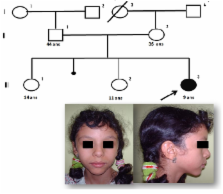
Les syndromes microdélétionnels sont définis par la présence d’une anomalie chromosomique de taille mineure (inférieure à 5 mégabases) ou aneusomie segmentaire, décelable par cytogénétique moléculaire (FISH : Fluorescent in Situ Hybridization). Les syndromes microdélétionnels représentent des syndromes cliniques avec des phénotypes suffisamment caractéristiques pour être reconnus cliniquement. Actuellement la FISH est la technique de choix pour rechercher ces syndromes. Plusieurs syndromes microdélétionnels peuvent être confirmés aisément, les plus recherchés sont Le syndrome de Williams (microdélétion en 7q11.23) et le syndrome de la délétion 22q11 (microdélétion en 22q11.2). Le syndrome de Williams est caractérisé par une anomalie du développement qui associe un retard psycho-moteur, une dysmorphie du visage évocatrice et un profil cognitif et comportemental spécifique, une sténose aortique supravalvulaire -SASV- le plus souvent. Le Syndrome de la délétion 22q11 se caractérise par l’association de plusieurs malformations d’expression variable: une cardiopathie congénitale de type conotroncal, une dysmorphie faciale discrète mais caractéristique et une hypoplasie du thymus et des parathyroïdes. Nous rapportons nos premières observations au CHU Hassan II confirmées par FISH : Syndrome de la délétion 22q11 (n:2) et un syndrome de Williams. Le but de cet article est la mise à jour de nos connaissances sur ces deux syndromes et la mise en valeur du rôle de la cytogénétique moléculaire dans le diagnostic et le conseil génétique des syndromes microdélétionnels.
Most cited references23
- Record: found
- Abstract: found
- Article: not found
Clinical features of 78 adults with 22q11 Deletion Syndrome.
- Record: found
- Abstract: not found
- Article: not found
Supravalvular aortic stenosis.
- Record: found
- Abstract: found
- Article: not found