- Record: found
- Abstract: found
- Article: found
MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data
Read this article at
Abstract
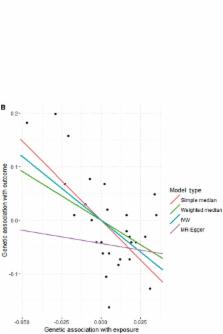
MendelianRandomization is a software package for the R open-source software environment that performs Mendelian randomization analyses using summarized data. The core functionality is to implement the inverse-variance weighted, MR-Egger and weighted median methods for multiple genetic variants. Several options are available to the user, such as the use of robust regression, fixed- or random-effects models and the penalization of weights for genetic variants with heterogeneous causal estimates. Extensions to these methods, such as allowing for variants to be correlated, can be chosen if appropriate. Graphical commands allow summarized data to be displayed in an interactive graph, or the plotting of causal estimates from multiple methods, for comparison. Although the main method of data entry is directly by the user, there is also an option for allowing summarized data to be incorporated from the PhenoScanner database of genotype—phenotype associations. We hope to develop this feature in future versions of the package. The R software environment is available for download from [ https://www.r-project.org/]. The MendelianRandomization package can be downloaded from the Comprehensive R Archive Network (CRAN) within R, or directly from [ https://cran.r-project.org/web/packages/MendelianRandomization/]. Both R and the MendelianRandomization package are released under GNU General Public Licenses (GPL-2|GPL-3).
Related collections
Most cited references8
- Record: found
- Abstract: found
- Article: not found
'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease?

- Record: found
- Abstract: found
- Article: found
Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic
- Record: found
- Abstract: found
- Article: not found