- Record: found
- Abstract: found
- Article: found
Heparanase mediates renal dysfunction during early sepsis in mice
Read this article at
Abstract
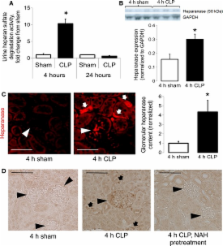
Heparanase, a heparan sulfate-specific glucuronidase, mediates the onset of pulmonary neutrophil adhesion and inflammatory lung injury during early sepsis. We hypothesized that glomerular heparanase is similarly activated during sepsis and contributes to septic acute kidney injury (AKI). We induced polymicrobial sepsis in mice using cecal ligation and puncture (CLP) in the presence or absence of competitive heparanase inhibitors (heparin or nonanticoagulant N-desulfated re- N-acetylated heparin [NAH]). Four hours after surgery, we collected serum and urine for measurement of renal function and systemic inflammation, invasively determined systemic hemodynamics, harvested kidneys for histology/protein/mRNA, and/or measured glomerular filtration by inulin clearance. CLP-treated mice demonstrated early activation of glomerular heparanase with coincident loss of glomerular filtration, as indicated by a >twofold increase in blood urea nitrogen (BUN) and a >50% decrease in inulin clearance ( P < 0.05) in comparison to sham mice. Administration of heparanase inhibitors 2 h prior to CLP attenuated sepsis-induced loss of glomerular filtration rate, demonstrating that heparanase activation contributes to early septic renal dysfunction. Glomerular heparanase activation was not associated with renal neutrophil influx or altered vascular permeability, in marked contrast to previously described effects of pulmonary heparanase on neutrophilic lung injury during sepsis. CLP induction of renal inflammatory gene (IL-6, TNF-α, IL-1β) expression was attenuated by NAH pretreatment. While serum inflammatory indices (KC, IL-6, TNF-α, IL-1β) were not impacted by NAH pretreatment, heparanase inhibition attenuated the CLP-induced increase in serum IL-10. These findings demonstrate that glomerular heparanase is active during sepsis and contributes to septic renal dysfunction via mechanisms disparate from heparanase-mediated lung injury.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Sepsis and acute kidney injury.
- Record: found
- Abstract: found
- Article: not found
Mechanisms of cardiac and renal dysfunction in patients dying of sepsis.
- Record: found
- Abstract: found
- Article: not found