- Record: found
- Abstract: found
- Article: found
Sequence-based prediction of protein protein interaction using a deep-learning algorithm
Read this article at
Abstract
Background
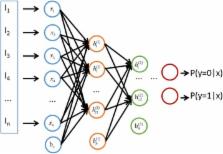
Protein-protein interactions (PPIs) are critical for many biological processes. It is therefore important to develop accurate high-throughput methods for identifying PPI to better understand protein function, disease occurrence, and therapy design. Though various computational methods for predicting PPI have been developed, their robustness for prediction with external datasets is unknown. Deep-learning algorithms have achieved successful results in diverse areas, but their effectiveness for PPI prediction has not been tested.
Results
We used a stacked autoencoder, a type of deep-learning algorithm, to study the sequence-based PPI prediction. The best model achieved an average accuracy of 97.19% with 10-fold cross-validation. The prediction accuracies for various external datasets ranged from 87.99% to 99.21%, which are superior to those achieved with previous methods.
Related collections
Most cited references28
- Record: found
- Abstract: found
- Article: not found
Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry.
- Record: found
- Abstract: found
- Article: not found
Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae.

- Record: found
- Abstract: found
- Article: found