- Record: found
- Abstract: found
- Article: found
Prediction of human-virus protein-protein interactions through a sequence embedding-based machine learning method

Read this article at
Graphical abstract
Highlights
Abstract
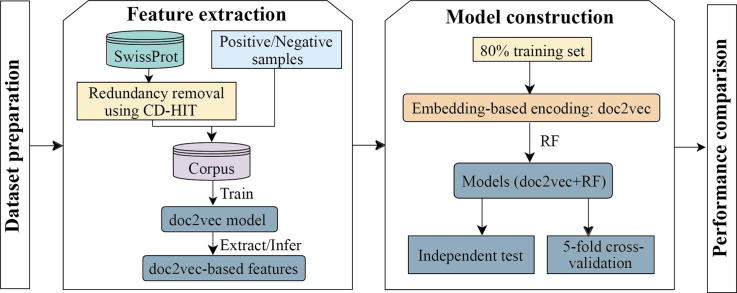
The identification of human-virus protein-protein interactions (PPIs) is an essential and challenging research topic, potentially providing a mechanistic understanding of viral infection. Given that the experimental determination of human-virus PPIs is time-consuming and labor-intensive, computational methods are playing an important role in providing testable hypotheses, complementing the determination of large-scale interactome between species. In this work, we applied an unsupervised sequence embedding technique (doc2vec) to represent protein sequences as rich feature vectors of low dimensionality. Training a Random Forest (RF) classifier through a training dataset that covers known PPIs between human and all viruses, we obtained excellent predictive accuracy outperforming various combinations of machine learning algorithms and commonly-used sequence encoding schemes. Rigorous comparison with three existing human-virus PPI prediction methods, our proposed computational framework further provided very competitive and promising performance, suggesting that the doc2vec encoding scheme effectively captures context information of protein sequences, pertaining to corresponding protein-protein interactions. Our approach is freely accessible through our web server as part of our host-pathogen PPI prediction platform ( http://zzdlab.com/InterSPPI/). Taken together, we hope the current work not only contributes a useful predictor to accelerate the exploration of human-virus PPIs, but also provides some meaningful insights into human-virus relationships.
Related collections
Most cited references41
- Record: found
- Abstract: found
- Article: not found
A comprehensive two-hybrid analysis to explore the yeast protein interactome.
- Record: found
- Abstract: found
- Article: not found
Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry.
- Record: found
- Abstract: found
- Article: not found