- Record: found
- Abstract: found
- Article: found
Gene expression profiling reveals novel TGFβ targets in adult lung fibroblasts
Read this article at
Abstract
Background
Transforming growth factor beta (TGFβ), a multifunctional cytokine, plays a crucial role in the accumulation of extracellular matrix components in lung fibrosis, where lung fibroblasts are considered to play a major role. Even though the effects of TGFβ on the gene expression of several proteins have been investigated in several lung fibroblast cell lines, the global pattern of response to this cytokine in adult lung fibroblasts is still unknown.
Methods
We used Affymetrix oligonucleotide microarrays U95v2, containing approximately 12,000 human genes, to study the transcriptional profile in response to a four hour treatment with TGFβ in control lung fibroblasts and in fibroblasts from patients with idiopathic and scleroderma-associated pulmonary fibrosis. A combination of the Affymetrix change algorithm (Microarray Suite 5) and of analysis of variance models was used to identify TGFβ-regulated genes. Additional criteria were an average up- or down- regulation of at least two fold.
Results
Exposure of fibroblasts to TGFβ had a profound impact on gene expression, resulting in regulation of 129 transcripts. We focused on genes not previously found to be regulated by TGFβ in lung fibroblasts or other cell types, including nuclear co-repressor 2, SMAD specific E3 ubiquitin protein ligase 2 ( SMURF2), bone morphogenetic protein 4, and angiotensin II receptor type 1 ( AGTR1), and confirmed the microarray results by real time-PCR. Western Blotting confirmed induction at the protein level of AGTR1, the most highly induced gene in both control and fibrotic lung fibroblasts among genes encoding for signal transduction molecules.
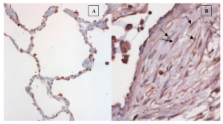
Upregulation of AGTR1 occurred through the MKK1/MKK2 signalling pathway. Immunohistochemical staining showed AGTR1 expression by lung fibroblasts in fibroblastic foci within biopsies of idiopathic pulmonary fibrosis.
Related collections
Most cited references40
- Record: found
- Abstract: found
- Article: not found
Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy.
- Record: found
- Abstract: found
- Article: not found
Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung.
- Record: found
- Abstract: found
- Article: not found