- Record: found
- Abstract: found
- Article: found
Origin and cross-species transmission of bat coronaviruses in China
Read this article at
Abstract
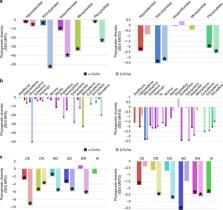
Bats are presumed reservoirs of diverse coronaviruses (CoVs) including progenitors of Severe Acute Respiratory Syndrome (SARS)-CoV and SARS-CoV-2, the causative agent of COVID-19. However, the evolution and diversification of these coronaviruses remains poorly understood. Here we use a Bayesian statistical framework and a large sequence data set from bat-CoVs (including 630 novel CoV sequences) in China to study their macroevolution, cross-species transmission and dispersal. We find that host-switching occurs more frequently and across more distantly related host taxa in alpha- than beta-CoVs, and is more highly constrained by phylogenetic distance for beta-CoVs. We show that inter-family and -genus switching is most common in Rhinolophidae and the genus Rhinolophus. Our analyses identify the host taxa and geographic regions that define hotspots of CoV evolutionary diversity in China that could help target bat-CoV discovery for proactive zoonotic disease surveillance. Finally, we present a phylogenetic analysis suggesting a likely origin for SARS-CoV-2 in Rhinolophus spp. bats.
Abstract
Bats are a likely reservoir of zoonotic coronaviruses (CoVs). Here, analyzing bat CoV sequences in China, the authors find that alpha-CoVs have switched hosts more frequently than betaCoVs, identify a bat family and genus that are highly involved in host-switching, and define hotspots of CoV evolutionary diversity.
Related collections
Most cited references48

- Record: found
- Abstract: found
- Article: found
A pneumonia outbreak associated with a new coronavirus of probable bat origin
- Record: found
- Abstract: found
- Article: not found
Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding
- Record: found
- Abstract: found
- Article: not found