- Record: found
- Abstract: found
- Article: found
In vivo HSC prime editing rescues sickle cell disease in a mouse model

Read this article at
Key Points
Visual Abstract
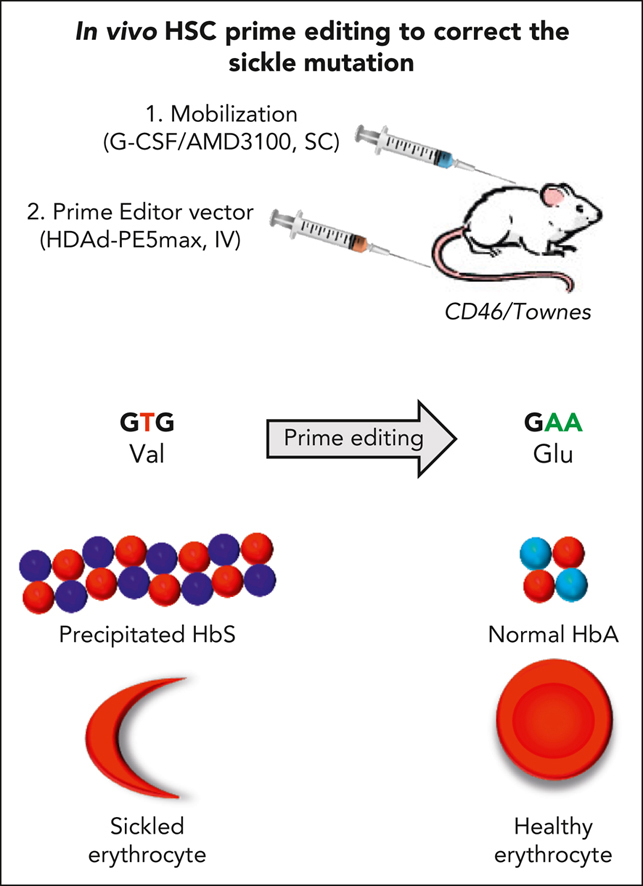
Abstract
Sickle cell disease (SCD) is a monogenic disease caused by a nucleotide mutation in the β-globin gene. Current gene therapy studies are mainly focused on lentiviral vector–mediated gene addition or CRISPR/Cas9–mediated fetal globin reactivation, leaving the root cause unfixed. We developed a vectorized prime editing system that can directly repair the SCD mutation in hematopoietic stem cells (HSCs) in vivo in a SCD mouse model (CD46/Townes mice). Our approach involved a single intravenous injection of a nonintegrating, prime editor–expressing viral vector into mobilized CD46/Townes mice and low-dose drug selection in vivo. This procedure resulted in the correction of ∼40% of β S alleles in HSCs. On average, 43% of sickle hemoglobin was replaced by adult hemoglobin, thereby greatly mitigating the SCD phenotypes. Transplantation in secondary recipients demonstrated that long-term repopulating HSCs were edited. Highly efficient target site editing was achieved with minimal generation of insertions and deletions and no detectable off-target editing. Because of its simplicity and portability, our in vivo prime editing approach has the potential for application in resource-poor countries where SCD is prevalent.
Abstract
Li and colleagues report on a novel gene-therapy approach to sickle cell disease. Rather than ex vivo manipulation with lentiviral gene addition or CRISPR/Cas9-mediated fetal hemoglobin reactivation, the authors describe injection of a nonintegrating prime editor–expressing vector into a sickle mouse model with correction of over 40% of hemoglobin S alleles in vivo. Though several features need to be optimized, this technique offers a potential for gene-therapy delivery in resource-poor settings.
Related collections
Most cited references60
- Record: found
- Abstract: found
- Article: not found
Search-and-replace genome editing without double-strand breaks or donor DNA
- Record: found
- Abstract: found
- Article: not found
CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia
- Record: found
- Abstract: found
- Article: not found