- Record: found
- Abstract: found
- Article: found
pIChemiSt — Free Tool for the Calculation of Isoelectric Points of Modified Peptides

Read this article at
Abstract
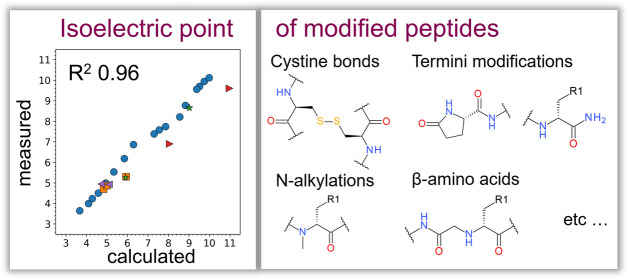
The isoelectric point (pI) is a fundamental physicochemical property of peptides and proteins. It is widely used to steer design away from low solubility and aggregation and guide peptide separation and purification. Experimental measurements of pI can be replaced by calculations knowing the ionizable groups of peptides and their corresponding p K a values. Different p K a sets are published in the literature for natural amino acids, however, they are insufficient to describe synthetically modified peptides, complex peptides of natural origin, and peptides conjugated with structures of other modalities. Noncanonical modifications (nCAAs) are ignored in the conventional sequence-based pI calculations, therefore producing large errors in their pI predictions. In this work, we describe a pI calculation method that uses the chemical structure as an input, automatically identifies ionizable groups of nCAAs and other fragments, and performs p K a predictions for them. The method is validated on a curated set of experimental measures on 29 modified and 119093 natural peptides, providing an improvement of R 2 from 0.74 to 0.95 and 0.96 against the conventional sequence-based approach for modified peptides for the two studied p K a prediction tools, ACDlabs and pKaMatcher, correspondingly. The method is available in the form of an open source Python library at https://github.com/AstraZeneca/peptide-tools, which can be integrated into other proprietary and free software packages. We anticipate that the pI calculation tool may facilitate optimization and purification activities across various application domains of peptides, including the development of biopharmaceuticals.
Related collections
Most cited references67
- Record: found
- Abstract: not found
- Article: not found
Matplotlib: A 2D Graphics Environment
- Record: found
- Abstract: found
- Article: not found
Biopython: freely available Python tools for computational molecular biology and bioinformatics
- Record: found
- Abstract: found
- Article: not found