- Record: found
- Abstract: found
- Article: found
A case study for cloud based high throughput analysis of NGS data using the globus genomics system

Read this article at
Abstract
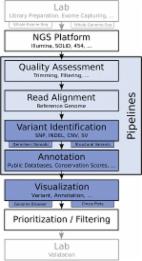
Next generation sequencing (NGS) technologies produce massive amounts of data requiring a powerful computational infrastructure, high quality bioinformatics software, and skilled personnel to operate the tools. We present a case study of a practical solution to this data management and analysis challenge that simplifies terabyte scale data handling and provides advanced tools for NGS data analysis. These capabilities are implemented using the “Globus Genomics” system, which is an enhanced Galaxy workflow system made available as a service that offers users the capability to process and transfer data easily, reliably and quickly to address end-to-endNGS analysis requirements. The Globus Genomics system is built on Amazon 's cloud computing infrastructure. The system takes advantage of elastic scaling of compute resources to run multiple workflows in parallel and it also helps meet the scale-out analysis needs of modern translational genomics research.
Related collections
Most cited references20

- Record: found
- Abstract: found
- Article: found
RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome
- Record: found
- Abstract: found
- Article: found