- Record: found
- Abstract: found
- Article: found
The importance of being connected: membrane contact sites and Parkinson’s disease
other
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Membrane contact sites (MCS) occur between closely apposed organelles and are a means
to transport ions and macromolecules between themselves, co-ordinate cellular metabolism,
and direct organelle fission and transport. While MCS between the endoplasmic reticulum
(ER) and mitochondria has long been investigated, the importance of MCS in both lipid
droplet (LD) function and the endolysosomal system are now being recognized. The identification
of VPS13C and LRRK2 at MCS, protein products of the familial Parkinson’s disease (PD)
loci PARK23 and PARK8, respectively, and the well-established dysfunction of the endolysosomal
system and mitochondria in disease pathogenesis, arguably put PD at the forefront
of MCS involvement in neurological disease.
A principal role of MCS is the transport of lipids and sterols between organelles.
The majority of lipids and sterols are synthesized in the ER and Golgi and trafficked
to endosomes, lysosomes, and the plasma membrane along the secretory pathway, with
a combination of localized lipid domains and specific cargo and sorting proteins targeting
them to the correct destination. Mitochondria are not on the secretory pathway, and
mitochondria-ER MCS sites are a way of delivering lipids. An example is the phospholipid
phosphatidylserine, which passes from the ER to mitochondria, and via enzymes located
on the inner mitochondrial membrane (IMM) is converted to phosphatidylethanolamine
and cardiolipin, two lipids that are crucial for the ultrastructure of cristae and
activity of the mitochondrial electron transport chain complexes (
Figure 1
). The VPS13 family of proteins has a hydrophobic core that can solubilize and transfer
glycerolipids between membranes. VPS13A and VPS13D are known to be at ER-mitochondria
MCS.
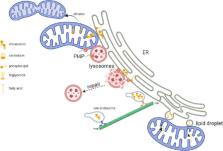
Figure 1
Membrane contact sites (MCS) between organelles.
Mitochondria rely on MCS with the endoplasmic reticulum (ER) to receive phospholipids
such as phosphatidylserine that can then be made into cardiolipin and phosphatidylethanolamine
in mitochondria, both of which aid oxidative phosphorylation and cristae morphology.
Mitochondria may also receive cholesterol and phosphatidylinositol-4-phosphate (PI4P)
from the ER and/or lysosomes, helping in mitochondrial DNA organization and division
of mitochondria. Damaged lysosomes form MCS with the ER, with PI4P facilitating the
transfer of cholesterol and other lipids from the ER to repair lysosomal membranes.
The cholesterol content of late endosomes dictates their cellular location, with high
cholesterol favoring association with motor dynein, thus moving them towards the negative
end of microtubules. Lipid droplet (LD) biogenesis occurs at ER and may also require
the presence of mitochondria. MCS between LD and mitochondria can channel fatty acids
liberated from triglycerides stored in LD straight into mitochondria, where it can
be used for β-oxidation. Created with BioRender.com.
Mitochondrial DNA (mtDNA) is organized into nucleoprotein complexes called nucleoids
and is associated with the IMM leaflet facing into the matrix. The complex associating
mtDNA to the IMM is thought to regulate the replication/organization of mtDNA and
its distribution (segregation) through the dynamic mitochondrial network. Mitochondrial
distribution depends on fission, which occurs at ER-mitochondria contact sites. It
is at these sites that nucleoids replicate and segregate. Cholesterol co-sediments
with mtDNA and disruption of cholesterol metabolism affect mtDNA organization, with
changes in the rigidity of the IMM a likely suspect (Desai et al., 2017).
An obvious source of nucleoid cholesterol would be the ER where it is synthesized.
However, lysosomes are also an important regulator of intracellular cholesterol, particularly
in neurons. In the adult brain, most brain cholesterol is synthesized in astrocytes
and exported to neurons via lipoproteins and processed via the endolysosomal system.
Export from late endosomes and lysosomes requires Niemann Pick C1 (NPC1) and NPC2
proteins. Mutations in NPC1 and NPC2 cause the neurological disorder Niemann Pick
type C, characterized by the accumulation of cholesterol and sphingolipids in the
lysosome. MCS between endosomes/lysosomes and the ER are thought to play a role in
the egress of cholesterol from the endocytic pathway, with NPC1 directly tethering
endosomes to the ER by interreacting with the ER sterol transport protein Gramd1b
(Höglinger et al., 2019). Notably, the same study found that cells with NPC1 or Gramd1b
depletion had an expanded population of lysosome-mitochondria MCS, via the late endosomal
sterol binding protein STARD3. These mitochondria had increased cholesterol levels.
Fibroblasts with NPC1 mutations exhibit distended mitochondria and perturbed mtDNA
organization and distribution (Desai et al., 2017).
Mitochondria and lysosomes both play a central role in regulating cellular metabolism,
with dysfunction in one organelle often affecting the other. Co-ordination between
these organelles is achieved by a variety of mechanisms including biogenesis, autophagy,
organelle movement/positioning, and modulation of enzyme activities, in anabolic and
catabolic pathways, to meet the needs of cells during for example nutrient deprivation
or a particular stress. The transcription factor TFEB is one player that is at the
heart of mitochondria-lysosome co-ordination. Two antagonistic metabolic hubs can
modify TFEB behavior, mTORC1, and AMPK. Active mTORC1 resides on the lysosome surface
and under high glucose and amino acids conditions keep TFEB located in the cytosol,
while also simultaneously inhibiting fatty acid (FA) oxidation and increasing lipogenesis
via the PPAR family. Low glucose or amino acids inactivates mTORC1, allowing TFEB
to move to the nucleus, stimulating lysosomal biogenesis and autophagy (to increase
nutrients) and the transcription of modulators of mitochondrial biogenesis such as
PGC-1α. mTORC1 inhibition also activates PPARα to increase β-oxidation of FA in mitochondria.
While lysosomal and mitochondrial function can be modulated via signaling cascades,
MCS between these organelles also occur. They have been estimated to occur in 15%
of the lysosomal population, lasting from seconds to minutes. In dopaminergic neurons,
these MCS occur at a similar rate in the soma, axons, and dendrites, but the duration
appeared longer in axons (Cisneros et al., 2022). The formation of mitochondria-lysosome
contacts is tightly regulated by Rab7. GTP-bound Rab7 promotes contact formation,
while GTP hydrolysis mediates untethering. Rab7 GTP hydrolysis is driven by TBC1D15,
which is recruited to the mitochondrial outer membrane by Fis1, an enzyme involved
in mitochondrial fission.
Variants in the GBA1 gene are the greatest genetic risk factor for developing PD.
Homozygote mutations cause the lysosomal storage disorder Gaucher disease. GBA1 encodes
the lysosomal enzyme glucocerebrosidase. Loss of glucocerebrosidase activity results
in lysosomal dysfunction, accumulation of α-synuclein, lipid dysregulation, and impaired
mitochondrial function. Dopaminergic neurons with GBA1 mutations exhibit prolonged
mitochondria-lysosome contacts, due to decreased TBC1D15, and could be reversed by
increasing glucocerebrosidase activity (Kim et al., 2021).
GDAP1 is a glutathione-S-transferase located on the mitochondrial outer membrane,
and in cultured neurons has been shown to form MCS with both the ER and lysosomes.
In the case of the latter, GDAP1 binds the endolysosomal integral membrane protein
LAMP1. Loss of GDAP1 activity resulted in mitochondrial dysfunction, enlarged lysosomes,
and delayed activation of TFEB (Cantarero et al., 2021). While it is difficult to
disentangle the direct effects of MCS from other events in the cells (such as autophagy
impairment), it once again highlights a potential ER-mitochondria-lysosome tripartite
assembly to help mediate mitochondrial fission. In another study, the ER was found
to recruit lysosomes to sites of mitochondrial fission via the interaction of the
VAMP-associated proteins with the lysosomal lipid transfer protein ORP1L. They proposed
that ORP1L transfers phosphatidylinositol-4-phosphate (PI4P) from lysosomes to mitochondria
increasing the efficiency of division (
Figure 1
).
VPS13C is found at MCS between the ER and late endosomes/lysosomes. Mutations in VPS13C
are a cause of early-onset autosomal recessive PD with loss of VPS13C function in
HeLa cells causing an accumulation of lysosomes with an altered lipid profile (Hancock-Cerutti
et al., 2022). This lysosomal dysfunction was implicated in the increase of the cGAS-STING
pathway seen in these cells. Aberrant activation of this immune pathway has been implicated
in PD pathogenesis.
ER-lysosomal MCS can activate mTORC1 on the lysosomal surface by increasing cholesterol
levels. Interaction of endosomes and lysosomes with the ER by MSC also affects their
cellular localization and movement. ER tubules are required for endosome fission.
Disruption of this process will affect the trafficking of newly synthesized enzymes
and proteins to the lysosome and affect the function of the retromer complex in the
opposite direction. ORP1L can act as a cholesterol sensor and in combination with
VAMP-associated proteins, Rab7 and Rab7-interacting lysosomal protein, modulate the
movement of late endosomes (
Figure 1
). Under high cholesterol levels, the complex recruits motors that drive the movement
towards the minus end of microtubules, resulting in a juxta-nuclear position. Conversely,
low cholesterol levels cause the recruitment of motors that will take endosomes to
the positive end (and periphery of the cell). Similarly in neurons, enlarged endolysosomes
with accumulation of intraluminal substrates have more MCS with ER, slowing movement
and increased clustering in the soma.
When lysosomes are damaged, the lysosomal membrane can become permeabilized, resulting
in the rapid accumulation of the enzyme phosphatidylinositol-4 kinase type 2α. This
increases PI4P levels on lysosomes, which consequently recruits oxysterol-binding
protein-related protein (ORP) family members such as ORP9, ORP10, ORP11, and oxysterol-binding
protein. This facilitates MCS between lysosomes and the ER, transferring phosphatidylserine
and cholesterol to help repair membranes. The lipid transporter ATG2 also helps directly
transfer lipids to lysosome membranes following activation by phosphatidylserine (Tan
and Finkel, 2022). Lysosomal membrane permeabilization can also cause lysosomes to
form tubules, which are then sorted into vesicles that then fuse with healthy lysosomes,
in a process termed lysosomal tubulation/sorting driven by LRRK2. As the name suggests
the PD protein LRRK2 promotes tubule formation phosphorylating Rab10, which then recruits
the motor adaptor protein JIP4. The fission of lysosomal tubules requires the binding
of tubular ER (Bonet-Ponce and Cookson, 2022).
LDs are organelles that allow the cell to store esterified lipids (e.g., triglycerides)
and sterols, allowing the cell a constant supply of lipids/sterols that is independent
of external nutrient availability. Furthermore, excess free FA can be toxic to cells
as they can become oxidized and initiate a damaging cascade of free radical damage.
The esterification of FA to triglycerides in the ER and storage in LD is a mechanism
by which cells, including neurons and glia, can reduce this risk. LD form MCS with
several organelles in their role as a hub in lipid metabolism. LD biogenesis occurs
at the ER, allowing direct flow of lipids and sterols made in the ER to the LD (
Figure 1
). Indeed, activation of the unfolded protein response and ER stress can stimulate
LD biogenesis as a means to store lipids while lipid synthesis pathways in the ER
are suspended. LDs have even been postulated to store unfolded proteins. The ER protein
seipin is important in regulating LD-ER contacts and LD biogenesis. VPS13A and VPS13C
are also found at ER-LD MCS. The presence of mitochondria in conjunction with ER at
sites of LD biogenesis is becoming increasingly recognized with the lipid transporters
mitoguardin-2, ORP5 and ORP8 all localized to this tripartite contact (Hong et al.,
2022).
Several functions of LD involve the perilipin (PLIN) family of proteins. Both the
biogenesis of LD and recruitment of lipases to LD to catabolize triglycerides back
into FA can be regulated by PLIN2 and PLIN3. The MCS between LD and mitochondria are
thought to be a means to directly channel “liberated” FA straight into mitochondria
for β-oxidation (
Figure 1
). Note that the transport of FA from mitochondria to LD during times of plenty has
also been suggested. PLIN1 and PLIN5 have both been implicated in these channeling
events. LD can also be degraded by macroautophagy, whereby LDs are engulfed into autophagosomes,
which then fuse with lysosomes for degradation of the contents. PLIN2 and PLIN3 are
also substrates for chaperone-mediated autophagy, adding another layer of control.
More direct contacts between LD and lysosomes have been proposed, although more in
yeast than mammalian systems. However, given the plethora of new MCS being recognized
between organelles, this is likely to change soon.
Given the lysosomal dysfunction observed in PD, it would be a surprise if more connections
between PD pathogenesis and LDs are not found, other than the direct link with VPS13C
mutants. Increased LD number has been observed in cells with GBA1 mutations (Smith
et al., 2023). The mechanism(s) remain unclear but could be via impairment of the
autophagy-lysosome pathway, lipid dyshomeostasis, induction of ER stress or mitochondrial
dysfunction – all pathways implicated in PD pathogenesis. Dysregulation of LD metabolism
has been connected with increased α-synuclein levels in neurons, while increased LD
number is associated with inflammation in glia, affecting phagocytosis and production
of proinflammatory molecules.
A cohort study has shown that 56% of PD patients have at least one putative damaging
variant in a lysosomal storage gene, including GBA1 and NPC1. The role of these proteins,
in addition to LRRK2 and VPS13C at MCS, either in a structural role or affecting their
regulation/formation, implicates MCS dysregulation in PD pathogenesis. This could
be manifested in both neurons and glia and contribute to hallmarks of PD such as abnormal
mtDNA copy number and other forms of mitochondrial dysfunction; impaired endolysosomal
function leading to α-synuclein accumulation and increased inflammatory pathways.
I would like to both thank and apologize to the many authors whose papers have contributed
greatly to this area of research, but due to restrictions on the number of references,
could not be cited.
Related collections
Most cited references10

- Record: found
- Abstract: found
- Article: found
NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress
D. Höglinger, T Burgoyne, E. Sanchez-Heras … (2019)
- Record: found
- Abstract: found
- Article: not found
A phosphoinositide signalling pathway mediates rapid lysosomal repair
Jay Xiaojun Tan, Toren Finkel (2022)

- Record: found
- Abstract: found
- Article: found
Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson’s disease
Soojin Kim, Yvette Wong, Fanding Gao … (2021)