- Record: found
- Abstract: found
- Article: found
Long-term safety and clinical outcomes of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency: two-year results
Read this article at
Abstract
Background
Olipudase alfa is a recombinant human acid sphingomyelinase (ASM) enzyme replacement therapy (ERT) for non-central-nervous-system manifestations of acid sphingomyelinase deficiency (ASMD). We report 2-year cumulative safety and efficacy data after olipudase alfa treatment in 20 children (four adolescents [12–17 year], nine children [6–11 year], and seven infants/early child [1–5 year]) with baseline splenomegaly and growth deficits who completed the 1-year ASCEND-Peds clinical trial (NCT02292654) and who continue to receive olipudase alfa in a long-term study (NCT02004704). Efficacy endpoints include spleen and liver volumes, diffusing capacity of the lung for carbon monoxide (DL CO), high-resolution computed tomography (HRCT) lung imaging, lipid profiles, liver function tests, and height Z-scores.
Results
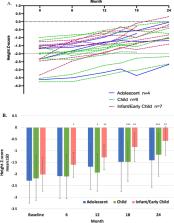
All 20 former ASCEND-Peds patients completed at least 2 years of olipudase alfa treatment. No patient discontinued and no new safety issue arose during the second year of treatment; 99% of adverse events were mild or moderate. During year 2, one patient had two treatment-related serious events of hypersensitivity that resolved. Mean reductions from baseline in spleen and liver volumes were 61% and 49%, respectively ( p < 0.0001) and mean percent-predicted-DL CO increased by 46.6% ( p < 0.0001) in nine patients who performed the test at baseline. Lipid profiles and elevated liver transaminase levels that improved or normalized by 1 year remained stable. Mean height Z-scores improved in all age groups (mean change from baseline 1.17, P < 0.0001).
Conclusion
Olipudase alfa was generally well-tolerated during 2 years of treatment. Improvements in clinically relevant disease endpoints observed during the first year of treatment were maintained or augmented in the second year.
Trial registration NCT02004704 registered 26 Nov 2013, https://clinicaltrials.gov/ct2/show/record/NCT02004704.
Related collections
Most cited references32
- Record: found
- Abstract: not found
- Article: not found
Interpretative strategies for lung function tests.
- Record: found
- Abstract: found
- Article: not found
Types A and B Niemann-Pick disease.
- Record: found
- Abstract: found
- Article: not found