- Record: found
- Abstract: found
- Article: found
Extensive metagenomic analysis of the porcine gut resistome to identify indicators reflecting antimicrobial resistance
Read this article at
Abstract
Background
Antimicrobial resistance (AMR) has been regarded as a major threat to global health. Pigs are considered an important source of antimicrobial resistance genes (ARGs). However, there is still a lack of large-scale quantitative data on the distribution of ARGs in the pig production industry. The bacterial species integrated ARGs in the gut microbiome have not been clarified.
Results
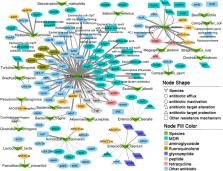
In the present study, we used deep metagenomic sequencing data of 451 samples from 425 pigs including wild boars, Tibetan pigs, and commercial or cross-bred experimental pigs under different rearing modes, to comprehensively survey the diversity and distribution of ARGs and detect the bacteria integrated in these ARGs. We identified a total of 1295 open reading frames (ORFs) recognized as antimicrobial resistance protein-coding genes. The ORFs were clustered into 349 unique types of ARGs, and these could be further classified into 69 drug resistance classes. Tetracycline resistance was most enriched in pig feces. Pigs raised on commercial farms had a significantly higher AMR level than pigs under semi-free ranging conditions or wild boars. We tracked the changes in the composition of ARGs at different growth stages and gut locations. There were 30 drug resistance classes showing significantly different abundances in pigs between 25 and 240 days of age. The richness of ARGs and 41 drug resistance classes were significantly different between cecum lumen and feces in pigs from commercial farms, but not in wild boars. We identified 24 bacterial species that existed in almost all tested samples (core bacteria) and were integrated 128 ARGs in their genomes. However, only nine ARGs of these 128 ARGs were core ARGs, suggesting that most of the ARGs in these bacterial species might be acquired rather than constitutive. We selected three subsets of ARGs as indicators for evaluating the pollution level of ARGs in samples with high accuracy ( r = 0.73~0.89).
Related collections
Most cited references66
- Record: found
- Abstract: found
- Article: not found
Fast gapped-read alignment with Bowtie 2.
- Record: found
- Abstract: found
- Article: not found
Cytoscape: a software environment for integrated models of biomolecular interaction networks.

- Record: found
- Abstract: found
- Article: found