- Record: found
- Abstract: found
- Article: not found
Assembly of the Dystrophin-Associated Protein Complex Does Not Require the Dystrophin Cooh-Terminal Domain
Read this article at

Abstract
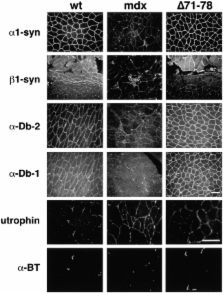
Dystrophin is a multidomain protein that links the actin cytoskeleton to laminin in the extracellular matrix through the dystrophin associated protein (DAP) complex. The COOH-terminal domain of dystrophin binds to two components of the DAP complex, syntrophin and dystrobrevin. To understand the role of syntrophin and dystrobrevin, we previously generated a series of transgenic mouse lines expressing dystrophins with deletions throughout the COOH-terminal domain. Each of these mice had normal muscle function and displayed normal localization of syntrophin and dystrobrevin. Since syntrophin and dystrobrevin bind to each other as well as to dystrophin, we have now generated a transgenic mouse deleted for the entire dystrophin COOH-terminal domain. Unexpectedly, this truncated dystrophin supported normal muscle function and assembly of the DAP complex. These results demonstrate that syntrophin and dystrobrevin functionally associate with the DAP complex in the absence of a direct link to dystrophin. We also observed that the DAP complexes in these different transgenic mouse strains were not identical. Instead, the DAP complexes contained varying ratios of syntrophin and dystrobrevin isoforms. These results suggest that alternative splicing of the dystrophin gene, which naturally generates COOH-terminal deletions in dystrophin, may function to regulate the isoform composition of the DAP complex.
Related collections
Most cited references88
- Record: found
- Abstract: found
- Article: not found
Dystrophin protects the sarcolemma from stresses developed during muscle contraction.
- Record: found
- Abstract: found
- Article: not found
Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix.
- Record: found
- Abstract: found
- Article: not found