- Record: found
- Abstract: found
- Article: found
Long-term maintenance of dystrophin expression and resistance to injury of skeletal muscle in gene edited DMD mice

Read this article at
Abstract
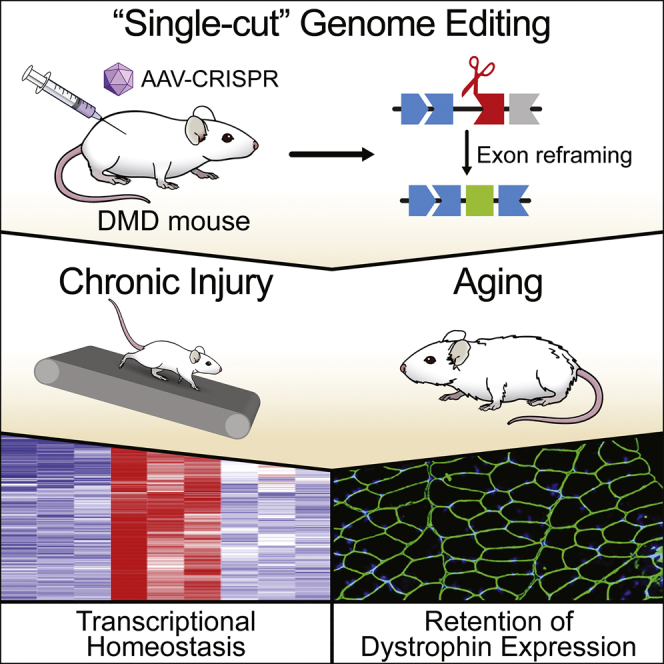
Duchenne muscular dystrophy (DMD) is a lethal muscle disease caused by mutations in the dystrophin gene. CRISPR/Cas9 genome editing has been used to correct DMD mutations in animal models at young ages. However, the longevity and durability of CRISPR/Cas9 editing remained to be determined. To address these issues, we subjected ΔEx44 DMD mice to systemic delivery of AAV9-expressing CRISPR/Cas9 gene editing components to reframe exon 45 of the dystrophin gene, allowing robust dystrophin expression and maintenance of muscle structure and function. We found that genome correction by CRISPR/Cas9 confers lifelong expression of dystrophin in mice and that corrected skeletal muscle is highly durable and resistant to myofiber necrosis and fibrosis, even in response to chronic injury. In contrast, when muscle fibers were ablated by barium chloride injection, we observed a loss of gene edited dystrophin expression. Analysis of on- and off-target editing in aged mice confirmed the stability of gene correction and the lack of significant off-target editing at 18 months of age. These findings demonstrate the long-term durability of CRISPR/Cas9 genome editing as a therapy for maintaining the integrity and function of DMD muscle, even under conditions of stress.
Graphical abstract
Abstract
CRISPR/Cas9 genome editing is used to correct Duchenne muscular dystrophy (DMD)-causing mutations. Here, Karri et al. demonstrate the durability and longevity of CRISPR/Cas9 genome editing in a mouse model of DMD, highlighting its potential as a long-term therapeutic for DMD patients.
Related collections
Most cited references46
- Record: found
- Abstract: found
- Article: not found
Search-and-replace genome editing without double-strand breaks or donor DNA
- Record: found
- Abstract: found
- Article: not found
Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy
- Record: found
- Abstract: found
- Article: not found