- Record: found
- Abstract: found
- Article: not found
Studies of Cobalt-Mediated Electrocatalytic CO 2 Reduction Using a Redox-Active Ligand
Read this article at

Abstract

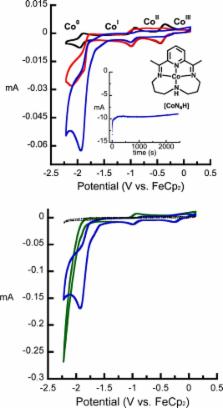
The cobalt complex [Co III N 4 H(Br) 2] + ( N 4 H = 2,12-dimethyl-3,7,11,17-tetraazabicyclo-[11.3.1]-heptadeca-1(7),2,11,13,15-pentaene) was used for electrocatalytic CO 2 reduction in wet MeCN with a glassy carbon working electrode. When water was employed as the proton source (10 M in MeCN), CO was produced ( f CO= 45% ± 6.4) near the Co I/0 redox couple for [Co III N 4 H(Br) 2] + ( E 1/2 = −1.88 V FeCp 2 +/0) with simultaneous H 2 evolution ( f H2= 30% ± 7.8). Moreover, we successfully demonstrated that the catalytically active species is homogeneous through the use of control experiments and XPS studies of the working glassy-carbon electrodes. As determined by cyclic voltammetry, CO 2 catalysis occurred near the formal Co I/0redox couple, and attempts were made to isolate the triply reduced compound (“[Co 0 N 4 H]”). Instead, the doubly reduced (“Co I”) compounds [Co N 4 ] and [Co N 4 H(MeCN)] + were isolated and characterized by X-ray crystallography. Their molecular structures prompted DFT studies to illuminate details regarding their electronic structure. The results indicate that reducing equivalents are stored on the ligand, implicating redox noninnocence in the ligands for H 2 evolution and CO 2 reduction electrocatalysis.
Abstract
Electrocatalytic CO 2 reduction was achieved using a homogeneous molecular cobalt complex in MeCN with 10 M H 2O using a glassy carbon working electrode. Control experiments and XPS measurements of the working electrode strongly suggest that the catalysis involves a molecular species rather than heterogeneous material. In addition to catalysis, stoichiometric reduction of the precatalyst resulted in the formation of several new reduced compounds in which the electronic structures were probed with XRD and DFT.
Related collections
Most cited references25
- Record: found
- Abstract: found
- Article: found
Rapid planetesimal formation in turbulent circumstellar discs
- Record: found
- Abstract: found
- Article: not found
Versatile Photocatalytic Systems for H2 Generation in Water Based on an Efficient DuBois-Type Nickel Catalyst
- Record: found
- Abstract: found
- Article: not found