- Record: found
- Abstract: found
- Article: found
Theoretical and Data-Driven Approaches for Biomolecular Condensates
Read this article at
Abstract
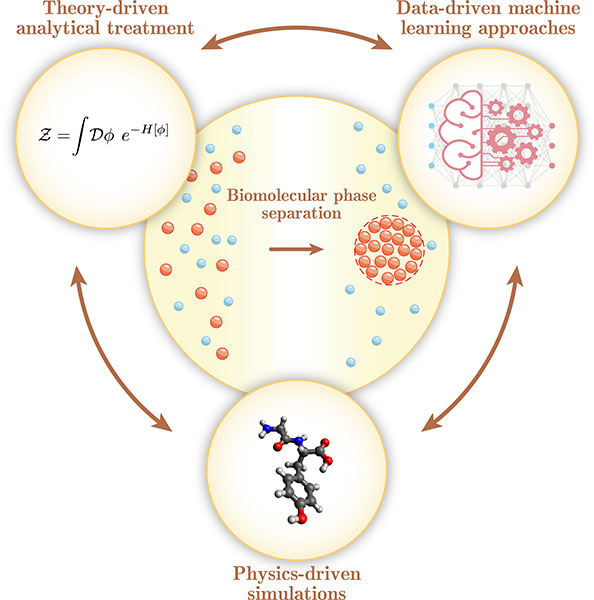
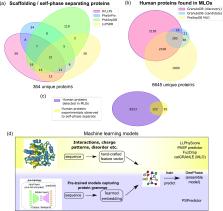
Biomolecular condensation processes are increasingly recognized as a fundamental mechanism that living cells use to organize biomolecules in time and space. These processes can lead to the formation of membraneless organelles that enable cells to perform distinct biochemical processes in controlled local environments, thereby supplying them with an additional degree of spatial control relative to that achieved by membrane-bound organelles. This fundamental importance of biomolecular condensation has motivated a quest to discover and understand the molecular mechanisms and determinants that drive and control this process. Within this molecular viewpoint, computational methods can provide a unique angle to studying biomolecular condensation processes by contributing the resolution and scale that are challenging to reach with experimental techniques alone. In this Review, we focus on three types of dry-lab approaches: theoretical methods, physics-driven simulations and data-driven machine learning methods. We review recent progress in using these tools for probing biomolecular condensation across all three fields and outline the key advantages and limitations of each of the approaches. We further discuss some of the key outstanding challenges that we foresee the community addressing next in order to develop a more complete picture of the molecular driving forces behind biomolecular condensation processes and their biological roles in health and disease.
Related collections
Most cited references202

- Record: found
- Abstract: found
- Article: found
Highly accurate protein structure prediction with AlphaFold
- Record: found
- Abstract: found
- Article: not found
CHARMM36m: an improved force field for folded and intrinsically disordered proteins
- Record: found
- Abstract: found
- Article: found