- Record: found
- Abstract: found
- Article: found
δ‐cells and β‐cells are electrically coupled and regulate α‐cell activity via somatostatin
Abstract
Key points
-
We used a mouse expressing a light‐sensitive ion channel in β‐cells to understand how α‐cell activity is regulated by β‐cells.
-
Light activation of β‐cells triggered a suppression of α‐cell activity via gap junction‐dependent activation of δ‐cells.
-
Mathematical modelling of human islets suggests that 23% of the inhibitory effect of glucose on glucagon secretion is mediated by β‐cells via gap junction‐dependent activation of δ‐cells/somatostatin secretion.
Abstract
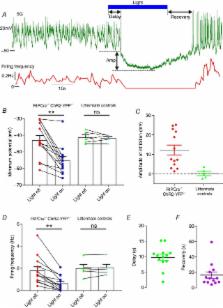
Glucagon, the body's principal hyperglycaemic hormone, is released from α‐cells of the pancreatic islet. Secretion of this hormone is dysregulated in type 2 diabetes mellitus but the mechanisms controlling secretion are not well understood. Regulation of glucagon secretion by factors secreted by neighbouring β‐ and δ‐cells (paracrine regulation) have been proposed to be important. In this study, we explored the importance of paracrine regulation by using an optogenetic strategy. Specific light‐induced activation of β‐cells in mouse islets expressing the light‐gated channelrhodopsin‐2 resulted in stimulation of electrical activity in δ‐cells but suppression of α‐cell activity. Activation of the δ‐cells was rapid and sensitive to the gap junction inhibitor carbenoxolone, whereas the effect on electrical activity in α‐cells was blocked by CYN 154806, an antagonist of the somatostatin‐2 receptor. These observations indicate that optogenetic activation of the β‐cells propagates to the δ‐cells via gap junctions, and the consequential stimulation of somatostatin secretion inhibits α‐cell electrical activity by a paracrine mechanism. To explore whether this pathway is important for regulating α‐cell activity and glucagon secretion in human islets, we constructed computational models of human islets. These models had detailed architectures based on human islets and consisted of a collection of >500 α‐, β‐ and δ‐cells. Simulations of these models revealed that this gap junctional/paracrine mechanism accounts for up to 23% of the suppression of glucagon secretion by high glucose.
Key points
-
We used a mouse expressing a light‐sensitive ion channel in β‐cells to understand how α‐cell activity is regulated by β‐cells.
-
Light activation of β‐cells triggered a suppression of α‐cell activity via gap junction‐dependent activation of δ‐cells.
-
Mathematical modelling of human islets suggests that 23% of the inhibitory effect of glucose on glucagon secretion is mediated by β‐cells via gap junction‐dependent activation of δ‐cells/somatostatin secretion.
Related collections
Most cited references78

- Record: found
- Abstract: found
- Article: not found
Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose
- Record: found
- Abstract: found
- Article: not found