- Record: found
- Abstract: found
- Article: found
The Therapeutic Potential of DNA Damage Repair Pathways and Genomic Stability in Lung Cancer
Read this article at
Abstract
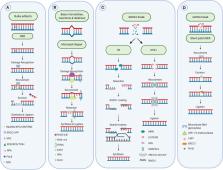
Despite advances in our understanding of the molecular biology of the disease and improved therapeutics, lung cancer remains the most common cause of cancer-related deaths worldwide. Therefore, an unmet need remains for improved treatments, especially in advanced stage disease. Genomic instability is a universal hallmark of all cancers. Many of the most commonly prescribed chemotherapeutics, including platinum-based compounds such as cisplatin, target the characteristic genomic instability of tumors by directly damaging the DNA. Chemotherapies are designed to selectively target rapidly dividing cells, where they cause critical DNA damage and subsequent cell death ( 1, 2). Despite the initial efficacy of these drugs, the development of chemotherapy resistant tumors remains the primary concern for treatment of all lung cancer patients. The correct functioning of the DNA damage repair machinery is essential to ensure the maintenance of normal cycling cells. Dysregulation of these pathways promotes the accumulation of mutations which increase the potential of malignancy. Following the development of the initial malignancy, the continued disruption of the DNA repair machinery may result in the further progression of metastatic disease. Lung cancer is recognized as one of the most genomically unstable cancers ( 3). In this review, we present an overview of the DNA damage repair pathways and their contributions to lung cancer disease occurrence and progression. We conclude with an overview of current targeted lung cancer treatments and their evolution toward combination therapies, including chemotherapy with immunotherapies and antibody-drug conjugates and the mechanisms by which they target DNA damage repair pathways.
Related collections
Most cited references107
- Record: found
- Abstract: found
- Article: not found
DNA double-strand breaks: signaling, repair and the cancer connection.
- Record: found
- Abstract: found
- Article: not found