- Record: found
- Abstract: found
- Article: found
A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data
Read this article at
Abstract
Motivation: The Illumina Infinium 450 k DNA Methylation Beadchip is a prime candidate technology for Epigenome-Wide Association Studies (EWAS). However, a difficulty associated with these beadarrays is that probes come in two different designs, characterized by widely different DNA methylation distributions and dynamic range, which may bias downstream analyses. A key statistical issue is therefore how best to adjust for the two different probe designs.
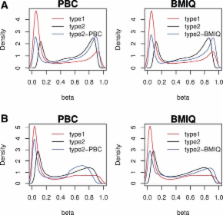
Results: Here we propose a novel model-based intra-array normalization strategy for 450 k data, called BMIQ (Beta MIxture Quantile dilation), to adjust the beta-values of type2 design probes into a statistical distribution characteristic of type1 probes. The strategy involves application of a three-state beta-mixture model to assign probes to methylation states, subsequent transformation of probabilities into quantiles and finally a methylation-dependent dilation transformation to preserve the monotonicity and continuity of the data. We validate our method on cell-line data, fresh frozen and paraffin-embedded tumour tissue samples and demonstrate that BMIQ compares favourably with two competing methods. Specifically, we show that BMIQ improves the robustness of the normalization procedure, reduces the technical variation and bias of type2 probe values and successfully eliminates the type1 enrichment bias caused by the lower dynamic range of type2 probes. BMIQ will be useful as a preprocessing step for any study using the Illumina Infinium 450 k platform.
Availability: BMIQ is freely available from http://code.google.com/p/bmiq/.
Contact: a.teschendorff@ 123456ucl.ac.uk
Supplementary information: Supplementary data are available at Bioinformatics online
Related collections
Most cited references18
- Record: found
- Abstract: found
- Article: not found
High density DNA methylation array with single CpG site resolution.
- Record: found
- Abstract: found
- Article: found
Epigenome-wide association studies for common human diseases.
- Record: found
- Abstract: found
- Article: not found