- Record: found
- Abstract: found
- Article: found
How accurately can one predict drug binding modes using AlphaFold models?
Read this article at
Abstract
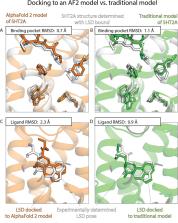
Computational prediction of protein structure has been pursued intensely for decades, motivated largely by the goal of using structural models for drug discovery. Recently developed machine-learning methods such as AlphaFold 2 (AF2) have dramatically improved protein structure prediction, with reported accuracy approaching that of experimentally determined structures. To what extent do these advances translate to an ability to predict more accurately how drugs and drug candidates bind to their target proteins? Here, we carefully examine the utility of AF2 protein structure models for predicting binding poses of drug-like molecules at the largest class of drug targets, the G-protein-coupled receptors. We find that AF2 models capture binding pocket structures much more accurately than traditional homology models, with errors nearly as small as differences between structures of the same protein determined experimentally with different ligands bound. Strikingly, however, the accuracy of ligand-binding poses predicted by computational docking to AF2 models is not significantly higher than when docking to traditional homology models and is much lower than when docking to structures determined experimentally without these ligands bound. These results have important implications for all those who might use predicted protein structures for drug discovery.
Related collections
Most cited references49

- Record: found
- Abstract: found
- Article: found
Highly accurate protein structure prediction with AlphaFold
- Record: found
- Abstract: found
- Article: not found
UCSF Chimera--a visualization system for exploratory research and analysis.
- Record: found
- Abstract: found
- Article: not found