- Record: found
- Abstract: found
- Article: not found
Crystal structure of the μ-opioid receptor bound to a morphinan antagonist
Read this article at

Summary
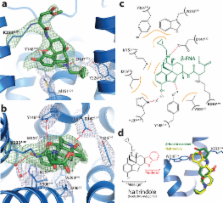
Opium is one of the world’s oldest drugs, and its derivatives morphine and codeine are among the most used clinical drugs to relieve severe pain. These prototypical opioids produce analgesia as well as many of their undesirable side effects (sedation, apnea and dependence) by binding to and activating the G-protein-coupled μ-opioid receptor (μOR) in the central nervous system. Here we describe the 2.8 Å crystal structure of the μOR in complex with an irreversible morphinan antagonist. Compared to the buried binding pocket observed in most GPCRs published to date, the morphinan ligand binds deeply within a large solvent-exposed pocket. Of particular interest, the μOR crystallizes as a two-fold symmetric dimer through a four-helix bundle motif formed by transmembrane segments 5 and 6. These high-resolution insights into opioid receptor structure will enable the application of structure-based approaches to develop better drugs for the management of pain and addiction.
Related collections
Most cited references51
- Record: found
- Abstract: found
- Article: not found
Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists.
- Record: found
- Abstract: found
- Article: not found
Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene.
- Record: found
- Abstract: found
- Article: not found