- Record: found
- Abstract: found
- Article: found
Genome‐wide DNA methylation analysis identifies potent CpG signature for temzolomide response in non‐G‐CIMP glioblastomas with unmethylated MGMT promoter: MGMT ‐dependent roles of GPR81

Read this article at
Abstract
Purposes
To identify potent DNA methylation candidates that could predict response to temozolomide (TMZ) in glioblastomas (GBMs) that do not have glioma‐CpGs island methylator phenotype (G‐CIMP) but have an unmethylated promoter of O‐6‐methylguanine‐DNA methyltransferase (un MGMT).
Methods
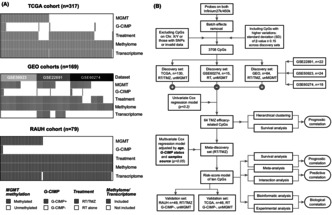
The discovery‐validation approach was planned incorporating a series of G‐CIMP−/un MGMT GBM cohorts with DNA methylation microarray data and clinical information, to construct multi‐CpG prediction models. Different bioinformatic and experimental analyses were performed for biological exploration.
Results
By analyzing discovery sets with radiotherapy (RT) plus TMZ versus RT alone, we identified a panel of 64 TMZ efficacy‐related CpGs, from which a 10‐CpG risk signature was further constructed. Both the 64‐CpG panel and the 10‐CpG risk signature were validated showing significant correlations with overall survival of G‐CIMP−/un MGMT GBMs when treated with RT/TMZ, rather than RT alone. The 10‐CpG risk signature was further observed for aiding TMZ choice by distinguishing differential outcomes to RT/TMZ versus RT within each risk subgroup. Functional studies on GPR81, the gene harboring one of the 10 CpGs, indicated its distinct impacts on TMZ resistance in GBM cells, which may be dependent on the status of MGMT expression.
Abstract
By analyzing DNA methylation microarray data and clinical information of glioblastomas (GBMs) that do not have glioma‐CpGs island methylator phenotype (G‐CIMP) but have an unmethylated promoter of O‐6‐methylguanine‐DNA methyltransferase (unMGMT), a panel of 64 CpGs and a 10‐CpG risk score signature were discovered and validated with specific linkage to temozolomide (TMZ) efficacy. Experimental data provided biological clues behind the risk signature. The 64 CpGs and in particular the 10‐CpG signature may serve as promising predictive biomarker candidates for guiding optimal usage of TMZ in GBMs with G‐CIMP‐ and un MGMT.
Related collections
Most cited references62
- Record: found
- Abstract: found
- Article: not found
Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles
- Record: found
- Abstract: found
- Article: not found
Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade.
- Record: found
- Abstract: found
- Article: not found