- Record: found
- Abstract: found
- Article: not found
Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death
brief-report

Scott J. Dixon
†
,
⊥
,
,
Georg E. Winter
∥ ,
Leila S. Musavi
† ,
Eric D. Lee
† ,
Berend Snijder
∥ ,
Manuele Rebsamen
∥ ,
Giulio Superti-Furga
∥
,
,
Brent R. Stockwell
†
,
‡
,
§
,
12 May 2015
Read this article at

There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
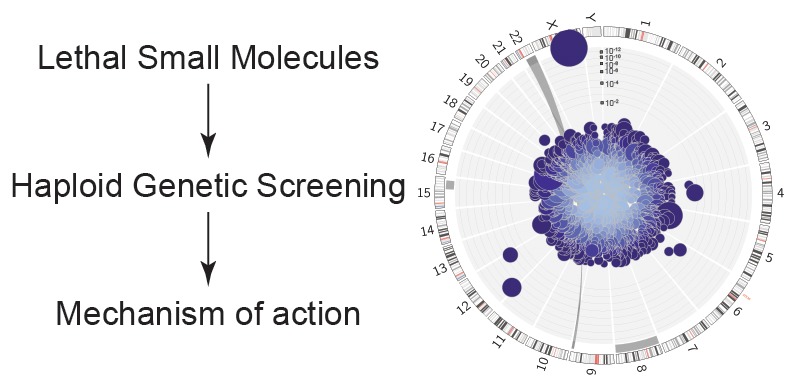
Little is known about the regulation of nonapoptotic cell death. Using massive insertional mutagenesis of haploid KBM7 cells we identified nine genes involved in small-molecule-induced nonapoptotic cell death, including mediators of fatty acid metabolism ( ACSL4) and lipid remodeling ( LPCAT3) in ferroptosis. One novel compound, CIL56, triggered cell death dependent upon the rate-limiting de novo lipid synthetic enzyme ACC1. These results provide insight into the genetic regulation of cell death and highlight the central role of lipid metabolism in nonapoptotic cell death.
Related collections
Most cited references16
- Record: found
- Abstract: found
- Article: not found
RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels.
- Record: found
- Abstract: found
- Article: not found
Synchronized renal tubular cell death involves ferroptosis.
Andreas Linkermann, Rachid Skouta, Nina Himmerkus … (2014)
- Record: found
- Abstract: found
- Article: not found
Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models
Rachid Skouta, Scott J. Dixon, Jianlin Wang … (2014)