- Record: found
- Abstract: found
- Article: found
CCM2–CCM3 interaction stabilizes their protein expression and permits endothelial network formation
Read this article at
Abstract
CCM2–CCM3 interactions protect CCM2 and CCM3 from proteasomal degradation, and both CCM2 and CCM3 are required for normal endothelial cell network formation.
Abstract
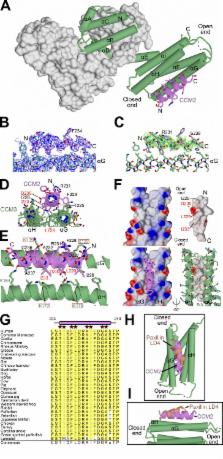
Mutations in the essential adaptor proteins CCM2 or CCM3 lead to cerebral cavernous malformations (CCM), vascular lesions that most frequently occur in the brain and are strongly associated with hemorrhagic stroke, seizures, and other neurological disorders. CCM2 binds CCM3, but the molecular basis of this interaction, and its functional significance, have not been elucidated. Here, we used x-ray crystallography and structure-guided mutagenesis to show that an α-helical LD-like motif within CCM2 binds the highly conserved “HP1” pocket of the CCM3 focal adhesion targeting (FAT) homology domain. By knocking down CCM2 or CCM3 and rescuing with binding-deficient mutants, we establish that CCM2–CCM3 interactions protect CCM2 and CCM3 proteins from proteasomal degradation and show that both CCM2 and CCM3 are required for normal endothelial cell network formation. However, CCM3 expression in the absence of CCM2 is sufficient to support normal cell growth, revealing complex-independent roles for CCM3.
Related collections
Most cited references25
- Record: found
- Abstract: found
- Article: not found
ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments.
- Record: found
- Abstract: found
- Article: not found
The molecular basis of filamin binding to integrins and competition with talin.
- Record: found
- Abstract: found
- Article: not found