- Record: found
- Abstract: found
- Article: found
ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules
Read this article at
Abstract
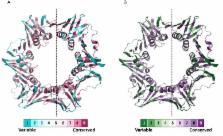
The degree of evolutionary conservation of an amino acid in a protein or a nucleic acid in DNA/RNA reflects a balance between its natural tendency to mutate and the overall need to retain the structural integrity and function of the macromolecule. The ConSurf web server ( http://consurf.tau.ac.il), established over 15 years ago, analyses the evolutionary pattern of the amino/nucleic acids of the macromolecule to reveal regions that are important for structure and/or function. Starting from a query sequence or structure, the server automatically collects homologues, infers their multiple sequence alignment and reconstructs a phylogenetic tree that reflects their evolutionary relations. These data are then used, within a probabilistic framework, to estimate the evolutionary rates of each sequence position. Here we introduce several new features into ConSurf, including automatic selection of the best evolutionary model used to infer the rates, the ability to homology-model query proteins, prediction of the secondary structure of query RNA molecules from sequence, the ability to view the biological assembly of a query (in addition to the single chain), mapping of the conservation grades onto 2D RNA models and an advanced view of the phylogenetic tree that enables interactively rerunning ConSurf with the taxa of a sub-tree.
Related collections
Most cited references48
- Record: found
- Abstract: found
- Article: not found
Dating of the human-ape splitting by a molecular clock of mitochondrial DNA.
- Record: found
- Abstract: found
- Article: not found
An evolutionary trace method defines binding surfaces common to protein families.
- Record: found
- Abstract: found
- Article: not found