- Record: found
- Abstract: found
- Article: found
Gasdermin D pore-forming activity is redox-sensitive
Read this article at
SUMMARY
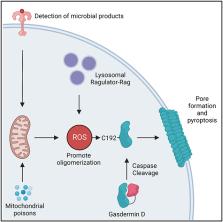
Reactive oxygen species (ROS) regulate the activities of inflammasomes, which are innate immune signaling organelles that induce pyroptosis. The mechanisms by which ROS control inflammasome activities are unclear and may be multifaceted. Herein, we report that the protein gasdermin D (GSDMD), which forms membrane pores upon cleavage by inflammasome-associated caspases, is a direct target of ROS. Exogenous and endogenous sources of ROS, and ROS-inducing stimuli that prime cells for pyroptosis induction, promote oligomerization of cleaved GSDMD, leading to membrane rupture and cell death. We find that ROS enhance GSDMD activities through oxidative modification of cysteine 192 (C192). Within macrophages, GSDMD mutants lacking C192 show impaired ability to form membrane pores and induce pyroptosis. Reciprocal mutagenesis studies reveal that C192 is the only cysteine within GSDMD that mediates ROS responsiveness. Cellular redox state is therefore a key determinant of GSDMD activities.
Graphical Abstract

In brief
Devant et al. investigate the actions of reactive oxygen species on the pore-forming protein GSDMD. They show that oxidation of a specific cysteine in GSDMD (C192) promotes its oligomerization, pore-forming activity, and pyroptosis. These findings reveal GSDMD as a redox-regulated protein that connects cellular redox state to inflammatory cell death.
Related collections
Most cited references41
- Record: found
- Abstract: found
- Article: not found
Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death.
- Record: found
- Abstract: found
- Article: not found
Reactive oxygen species (ROS) as pleiotropic physiological signalling agents
- Record: found
- Abstract: found
- Article: not found