- Record: found
- Abstract: found
- Article: found
PEAR: a fast and accurate Illumina Paired-End reAd mergeR
Read this article at
Abstract
Motivation: The Illumina paired-end sequencing technology can generate reads from both ends of target DNA fragments, which can subsequently be merged to increase the overall read length. There already exist tools for merging these paired-end reads when the target fragments are equally long. However, when fragment lengths vary and, in particular, when either the fragment size is shorter than a single-end read, or longer than twice the size of a single-end read, most state-of-the-art mergers fail to generate reliable results. Therefore, a robust tool is needed to merge paired-end reads that exhibit varying overlap lengths because of varying target fragment lengths.
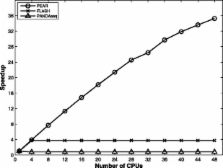
Results: We present the PEAR software for merging raw Illumina paired-end reads from target fragments of varying length. The program evaluates all possible paired-end read overlaps and does not require the target fragment size as input. It also implements a statistical test for minimizing false-positive results. Tests on simulated and empirical data show that PEAR consistently generates highly accurate merged paired-end reads. A highly optimized implementation allows for merging millions of paired-end reads within a few minutes on a standard desktop computer. On multi-core architectures, the parallel version of PEAR shows linear speedups compared with the sequential version of PEAR.
Availability and implementation: PEAR is implemented in C and uses POSIX threads. It is freely available at http://www.exelixis-lab.org/web/software/pear.
Contact: Tomas.Flouri@ 123456h-its.org
Related collections
Most cited references19
- Record: found
- Abstract: found
- Article: not found
Fast gapped-read alignment with Bowtie 2.
- Record: found
- Abstract: found
- Article: not found
FLASH: fast length adjustment of short reads to improve genome assemblies.
- Record: found
- Abstract: found
- Article: not found