- Record: found
- Abstract: found
- Article: not found
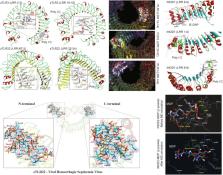
Structure of fish Toll-like receptors (TLR) and NOD-like receptors (NLR)
Read this article at

Abstract
Innate immunity driven by pattern recognition receptor (PRR) protects the host from invading pathogens. Aquatic animals like fish where the adaptive immunity is poorly developed majorly rely on their innate immunity modulated by PRRs like toll-like receptors (TLR) and NOD-like receptors (NLR). However, current development to improve the fish immunity via TLR/NLR signaling is affected by a poor understanding of its mechanistic and structural features. This review discusses the structure of fish TLRs/NLRs and its interaction with pathogen associated molecular patterns (PAMPs) and downstream signaling molecules. Over the past one decade, significant progress has been done in studying the structure of TLRs/NLRs in higher eukaryotes; however, structural studies on fish innate immune receptors are undermined. Several novel TLR genes are identified in fish that are absent in higher eukaryotes, but the function is still poorly understood. Unlike the fundamental progress achieved in developing antagonist/agonist to modulate human innate immunity, analogous studies in fish are nearly lacking due to structural inadequacy. This underlies the importance of exploring the structural and mechanistic details of fish TLRs/NLRs at an atomic and molecular level. This review outlined the mechanistic and structural basis of fish TLR and NLR activation.
Related collections
Most cited references100
- Record: found
- Abstract: found
- Article: not found
Improved protein-ligand docking using GOLD.
- Record: found
- Abstract: found
- Article: not found
Molecular Dynamics Simulation for All
- Record: found
- Abstract: found
- Article: not found