- Record: found
- Abstract: found
- Article: found
Measuring the tail: Methods for poly(A) tail profiling

Read this article at
Abstract
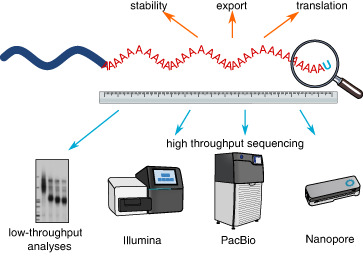
The 3′‐end poly(A) tail is an important and potent feature of most mRNA molecules that affects mRNA fate and translation efficiency. Polyadenylation is a posttranscriptional process that occurs in the nucleus by canonical poly(A) polymerases (PAPs). In some specific instances, the poly(A) tail can also be extended in the cytoplasm by noncanonical poly(A) polymerases (ncPAPs). This epitranscriptomic regulation of mRNA recently became one of the most interesting aspects in the field. Advances in RNA sequencing technologies and software development have allowed the precise measurement of poly(A) tails, identification of new ncPAPs, expansion of the function of known enzymes, discovery and a better understanding of the physiological role of tail heterogeneity, and recognition of a correlation between tail length and RNA translatability. Here, we summarize the development of polyadenylation research methods, including classic low‐throughput approaches, Illumina‐based genome‐wide analysis, and advanced state‐of‐art techniques that utilize long‐read third‐generation sequencing with Pacific Biosciences and Oxford Nanopore Technologies platforms. A boost in technical opportunities over recent decades has allowed a better understanding of the regulation of gene expression at the mRNA level.
This article is categorized under:
Abstract
Related collections
Most cited references138
- Record: found
- Abstract: found
- Article: not found
Full-length RNA-seq from single cells using Smart-seq2.
- Record: found
- Abstract: found
- Article: not found
The Architecture of SARS-CoV-2 Transcriptome
- Record: found
- Abstract: found
- Article: not found