- Record: found
- Abstract: found
- Article: found
Centronuclear (myotubular) myopathy
Read this article at
Abstract
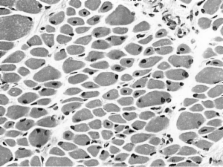
Centronuclear myopathy (CNM) is an inherited neuromuscular disorder characterised by clinical features of a congenital myopathy and centrally placed nuclei on muscle biopsy.
The incidence of X-linked myotubular myopathy is estimated at 2/100000 male births but epidemiological data for other forms are not currently available.
The clinical picture is highly variable. The X-linked form usually gives rise to a severe phenotype in males presenting at birth with marked weakness and hypotonia, external ophthalmoplegia and respiratory failure. Signs of antenatal onset comprise reduced foetal movements, polyhydramnios and thinning of the ribs on chest radiographs; birth asphyxia may be the present. Affected infants are often macrosomic, with length above the 90 th centile and large head circumference. Testes are frequently undescended. Both autosomal-recessive (AR) and autosomal-dominant (AD) forms differ from the X-linked form regarding age at onset, severity, clinical characteristics and prognosis. In general, AD forms have a later onset and milder course than the X-linked form, and the AR form is intermediate in both respects.
Mutations in the myotubularin (MTM1) gene on chromosome Xq28 have been identified in the majority of patients with the X-linked recessive form, whilst AD and AR forms have been associated with mutations in the dynamin 2 (DNM2) gene on chromosome 19p13.2 and the amphiphysin 2 (BIN1) gene on chromosome 2q14, respectively. Single cases with features of CNM have been associated with mutations in the skeletal muscle ryanodine receptor (RYR1) and the hJUMPY (MTMR14) genes.
Diagnosis is based on typical histopathological findings on muscle biopsy in combination with suggestive clinical features; muscle magnetic resonance imaging may complement clinical assessment and inform genetic testing in cases with equivocal features. Genetic counselling should be offered to all patients and families in whom a diagnosis of CNM has been made.
The main differential diagnoses include congenital myotonic dystrophy and other conditions with severe neonatal hypotonia.
Management of CNM is mainly supportive, based on a multidisciplinary approach. Whereas the X-linked form due to MTM1 mutations is often fatal in infancy, dominant forms due to DNM2 mutations and some cases of the recessive BIN1-related form appear to be associated with an overall more favourable prognosis.
Related collections
Most cited references144
- Record: found
- Abstract: found
- Article: not found
BAR domains as sensors of membrane curvature: the amphiphysin BAR structure.
- Record: found
- Abstract: found
- Article: not found
Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle.
- Record: found
- Abstract: found
- Article: not found