- Record: found
- Abstract: found
- Article: found
Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases
Read this article at
Abstract
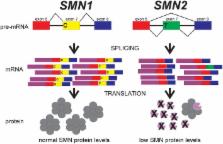
Proximal spinal muscular atrophy (SMA), a leading genetic cause of infant death worldwide, is an early-onset, autosomal recessive neurodegenerative disease characterized by the loss of spinal α-motor neurons. This loss of α-motor neurons is associated with muscle weakness and atrophy. SMA can be classified into five clinical grades based on age of onset and severity of the disease. Regardless of clinical grade, proximal SMA results from the loss or mutation of SMN1 ( survival motor neuron 1) on chromosome 5q13. In humans a large tandem chromosomal duplication has lead to a second copy of the SMN gene locus known as SMN2. SMN2 is distinguishable from SMN1 by a single nucleotide difference that disrupts an exonic splice enhancer in exon 7. As a result, most of SMN2 mRNAs lack exon 7 ( SMNΔ 7) and produce a protein that is both unstable and less than fully functional. Although only 10–20% of the SMN2 gene product is fully functional, increased genomic copies of SMN2 inversely correlates with disease severity among individuals with SMA. Because SMN2 copy number influences disease severity in SMA, there is prognostic value in accurate measurement of SMN2 copy number from patients being evaluated for SMA. This prognostic value is especially important given that SMN2 copy number is now being used as an inclusion criterion for SMA clinical trials. In addition to SMA, copy number variations (CNVs) in the SMN genes can affect the clinical severity of other neurological disorders including amyotrophic lateral sclerosis (ALS) and progressive muscular atrophy (PMA). This review will discuss how SMN1 and SMN2 CNVs are detected and why accurate measurement of SMN1 and SMN2 copy numbers is relevant for SMA and other neurodegenerative diseases.
Related collections
Most cited references100
- Record: found
- Abstract: found
- Article: not found
SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing.
- Record: found
- Abstract: found
- Article: not found