- Record: found
- Abstract: found
- Article: not found
A new approach for determining SARS-CoV-2 epitopes using machine learning-based in silico methods
Read this article at

Abstract
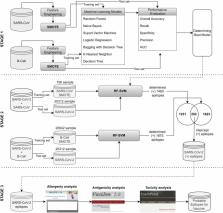
The emergence of machine learning-based in silico tools has enabled rapid and high-quality predictions in the biomedical field. In the COVID-19 pandemic, machine learning methods have been used in many topics such as predicting the death of patients, modeling the spread of infection, determining future effects, diagnosis with medical image analysis, and forecasting the vaccination rate. However, there is a gap in the literature regarding identifying epitopes that can be used in fast, useful, and effective vaccine design using machine learning methods and bioinformatics tools. Machine learning methods can give medical biotechnologists an advantage in designing a faster and more successful vaccine. The motivation of this study is to propose a successful hybrid machine learning method for SARS-CoV-2 epitope prediction and to identify nonallergen, nontoxic, antigen peptides that can be used in vaccine design from the predicted epitopes with bioinformatics tools. The identified epitopes will be effective not only in the design of the COVID-19 vaccine but also against viruses from the SARS family that may be encountered in the future. For this purpose, epitope prediction performances of random forest, support vector machine, logistic regression, bagging with decision tree, k-nearest neighbor and decision tree methods were examined. In the SARS-CoV and B-cell datasets used for education in the study, epitope estimation was performed again after the datasets were balanced with the synthetic minority oversampling technique (SMOTE) method since the epitope class samples were in the minority compared to the nonepitope class. The experimental results obtained were compared and the most successful predictions were obtained with the random forest (RF) method. The epitope prediction performance in balanced datasets was found to be higher than that in the original datasets (94.0% AUC and 94.4% PRC for the SMOTE-SARS-CoV dataset; 95.6% AUC and 95.3% PRC for the SMOTE-B-cell dataset). In this study, 252 peptides out of 20312 peptides were determined to be epitopes with the SMOTE-RF-SVM hybrid method proposed for SARS-CoV-2 epitope prediction. Determined epitopes were analyzed with AllerTOP 2.0, VaxiJen 2.0 and ToxinPred tools, and allergic, nonantigen, and toxic epitopes were eliminated. As a result, 11 possible nonallergic, high antigen and nontoxic epitope candidates were proposed that could be used in protein-based COVID-19 vaccine design (“VGGNYNY”, “VNFNFNGLTG”, “RQIAPGQTGKI”, “QIAPGQTGKIA”, “SYECDIPIGAGI”, “STFKCYGVSPTKL”, “GVVFLHVTYVPAQ”, “KNHTSPDVDLGDI”, “NHTSPDVDLGDIS”, “AGAAAYYVGYLQPR”, “KKSTNLVKNKCVNF”). It is predicted that the few epitopes determined by machine learning-based in silico methods will help biotechnologists design fast and accurate vaccines by reducing the number of trials in the laboratory environment.
Graphical abstract

Related collections
Most cited references58
- Record: found
- Abstract: found
- Article: not found
A Novel Coronavirus from Patients with Pneumonia in China, 2019

- Record: found
- Abstract: found
- Article: found
A pneumonia outbreak associated with a new coronavirus of probable bat origin
- Record: found
- Abstract: found
- Article: not found