- Record: found
- Abstract: found
- Article: not found
RBM3 mediates structural plasticity and protective effects of cooling in neurodegeneration
Read this article at

Abstract
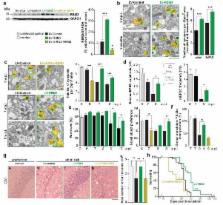
In the healthy adult brain synapses are continuously remodelled through a process of elimination and formation known as structural plasticity 1 . Reduction in synapse number is a consistent early feature of neurodegenerative diseases 2, 3 , suggesting deficient compensatory mechanisms. While much is known about toxic processes leading to synaptic dysfunction and loss in these disorders 2, 3 , how synaptic regeneration is affected is unknown. In hibernating mammals, cooling induces loss of synaptic contacts, which are reformed on rewarming, a form of structural plasticity 4, 5 . We have found that similar changes occur in artificially cooled laboratory rodents. Cooling and hibernation also induce a number cold-shock proteins in the brain, including the RNA binding protein, RBM3 6 . The relationship of such proteins to structural plasticity is unknown. Here we show that synapse regeneration is impaired in mouse models of neurodegenerative disease, in association with the failure to induce RBM3. In both prion-infected and 5×FAD (Alzheimer-type) mice 7 , the capacity to regenerate synapses after cooling declined in parallel with the loss of induction of RBM3. Enhanced expression of RBM3 in the hippocampus prevented this deficit and restored the capacity for synapse reassembly after cooling. Further, RBM3 over-expression, achieved either by boosting endogenous levels through hypothermia prior to the loss of the RBM3 response, or by lentiviral delivery, resulted in sustained synaptic protection in 5×FAD mice and throughout the course of prion disease, preventing behavioural deficits and neuronal loss and significantly prolonging survival. In contrast, knockdown of RBM3 exacerbated synapse loss in both models and accelerated disease and prevented the neuroprotective effects of cooling. Thus, deficient synapse regeneration, mediated at least in part by failure of the RBM3 stress response, contributes to synapse loss throughout the course of neurodegenerative disease. The data support enhancing cold shock pathways as potential protective therapies in neurodegenerative disorders.
Related collections
Most cited references30
- Record: found
- Abstract: found
- Article: not found
Sustained translational repression by eIF2α-P mediates prion neurodegeneration.
- Record: found
- Abstract: found
- Article: not found
Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice.
- Record: found
- Abstract: found
- Article: not found