- Record: found
- Abstract: found
- Article: found
Replication fork regression in vitro by the Werner syndrome protein (WRN): Holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity
Read this article at
Abstract
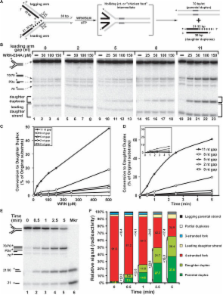
The premature aging and cancer-prone disease Werner syndrome stems from loss of WRN protein function. WRN deficiency causes replication abnormalities, sensitivity to certain genotoxic agents, genomic instability and early replicative senescence in primary fibroblasts. As a RecQ helicase family member, WRN is a DNA-dependent ATPase and unwinding enzyme, but also possesses strand annealing and exonuclease activities. RecQ helicases are postulated to participate in pathways responding to replication blockage, pathways possibly initiated by fork regression. In this study, a series of model replication fork substrates were used to examine the fork regression capability of WRN. Our results demonstrate that WRN catalyzes fork regression and Holliday junction formation. This process is an ATP-dependent reaction that is particularly efficient on forks containing single-stranded gaps of at least 11–13 nt on the leading arm at the fork junction. Importantly, WRN exonuclease activity, by digesting the leading daughter strand, enhances regression of forks with smaller gaps on the leading arm, thus creating an optimal structure for regression. Our results suggest that the multiple activities of WRN cooperate to promote replication fork regression. These findings, along with the established cellular consequences of WRN deficiency, strongly support a role for WRN in regression of blocked replication forks.
Related collections
Most cited references63
- Record: found
- Abstract: found
- Article: not found
The Bloom's syndrome helicase suppresses crossing over during homologous recombination.
- Record: found
- Abstract: found
- Article: not found
The Bloom's syndrome gene product is homologous to RecQ helicases.
- Record: found
- Abstract: found
- Article: not found