- Record: found
- Abstract: found
- Article: found
Loss of Protein Kinase Novel 1 (PKN1) is associated with mild systolic and diastolic contractile dysfunction, increased phospholamban Thr 17 phosphorylation, and exacerbated ischaemia-reperfusion injury
Abstract
Aims
PKN1 is a stress-responsive protein kinase acting downstream of small GTP-binding proteins of the Rho/Rac family. The aim was to determine its role in endogenous cardioprotection.
Methods and results
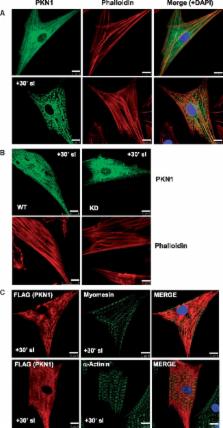
Hearts from PKN1 knockout (KO) or wild type (WT) littermate control mice were perfused in Langendorff mode and subjected to global ischaemia and reperfusion (I/R). Myocardial infarct size was doubled in PKN1 KO hearts compared to WT hearts. PKN1 was basally phosphorylated on the activation loop Thr 778 PDK1 target site which was unchanged during I/R. However, phosphorylation of p42/p44-MAPK was decreased in KO hearts at baseline and during I/R. In cultured neonatal rat ventricular cardiomyocytes (NRVM) and NRVM transduced with kinase dead (KD) PKN1 K 644R mutant subjected to simulated ischaemia/reperfusion (sI/R), PhosTag ® gel analysis showed net dephosphorylation of PKN1 during sI and early R despite Thr 778 phosphorylation. siRNA knockdown of PKN1 in NRVM significantly decreased cell survival and increased cell injury by sI/R which was reversed by WT- or KD-PKN1 expression. Confocal immunofluorescence analysis of PKN1 in NRVM showed increased localization to the sarcoplasmic reticulum (SR) during sI. GC-MS/MS and immunoblot analysis of PKN1 immunoprecipitates following sI/R confirmed interaction with CamKIIδ. Co-translocation of PKN1 and CamKIIδ to the SR/membrane fraction during sI correlated with phospholamban (PLB) Thr 17 phosphorylation. siRNA knockdown of PKN1 in NRVM resulted in increased basal CamKIIδ activation and increased PLB Thr 17 phosphorylation only during sI. In vivo PLB Thr 17 phosphorylation, Sarco-Endoplasmic Reticulum Ca 2+ ATPase (SERCA2) expression and Junctophilin-2 (Jph2) expression were also basally increased in PKN1 KO hearts. Furthermore, in vivo P-V loop analysis of the beat-to-beat relationship between rate of LV pressure development or relaxation and end diastolic P (EDP) showed mild but significant systolic and diastolic dysfunction with preserved ejection fraction in PKN1 KO hearts.
Conclusion
Loss of PKN1 in vivo significantly reduces endogenous cardioprotection and increases myocardial infarct size following I/R injury. Cardioprotection by PKN1 is associated with reduced CamKIIδ-dependent PLB Thr 17 phosphorylation at the SR and therefore may stabilize the coupling of SR Ca 2+ handling and contractile function, independent of its kinase activity.
Related collections
Most cited references46
- Record: found
- Abstract: found
- Article: not found
The extended protein kinase C superfamily.
- Record: found
- Abstract: found
- Article: not found
CaMKII determines mitochondrial stress responses in heart
- Record: found
- Abstract: found
- Article: not found