- Record: found
- Abstract: found
- Article: found
A New Method for Predicting the Subcellular Localization of Eukaryotic Proteins with Both Single and Multiple Sites: Euk-mPLoc 2.0

Read this article at
Abstract
Information of subcellular locations of proteins is important for in-depth studies
of cell biology. It is very useful for proteomics, system biology and drug development
as well. However, most existing methods for predicting protein subcellular location
can only cover 5 to 12 location sites. Also, they are limited to deal with single-location
proteins and hence failed to work for multiplex proteins, which can simultaneously
exist at, or move between, two or more location sites. Actually, multiplex proteins
of this kind usually posses some important biological functions worthy of our special
notice. A new predictor called “
Euk-mPLoc 2.0” is developed by hybridizing the gene ontology information, functional domain information,
and sequential evolutionary information through three different modes of pseudo amino
acid composition. It can be used to identify eukaryotic proteins among the following
22 locations: (1) acrosome, (2) cell wall, (3) centriole, (4) chloroplast, (5) cyanelle,
(6) cytoplasm, (7) cytoskeleton, (8) endoplasmic reticulum, (9) endosome, (10) extracell,
(11) Golgi apparatus, (12) hydrogenosome, (13) lysosome, (14) melanosome, (15) microsome
(16) mitochondria, (17) nucleus, (18) peroxisome, (19) plasma membrane, (20) plastid,
(21) spindle pole body, and (22) vacuole. Compared with the existing methods for predicting
eukaryotic protein subcellular localization, the new predictor is much more powerful
and flexible, particularly in dealing with proteins with multiple locations and proteins
without available accession numbers. For a newly-constructed stringent benchmark dataset
which contains both single- and multiple-location proteins and in which none of proteins
has
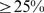 pairwise sequence identity to any other in a same location, the overall jackknife
success rate achieved by
Euk-mPLoc 2.0 is more than 24% higher than those by any of the existing predictors. As a user-friendly
web-server, Euk-mPLoc 2.0 is freely accessible at
http://www.csbio.sjtu.edu.cn/bioinf/euk-multi-2/. For a query protein sequence of 400 amino acids, it will take about 15 seconds for
the web-server to yield the predicted result; the longer the sequence is, the more
time it may usually need. It is anticipated that the novel approach and the powerful
predictor as presented in this paper will have a significant impact to Molecular Cell
Biology, System Biology, Proteomics, Bioinformatics, and Drug Development.
pairwise sequence identity to any other in a same location, the overall jackknife
success rate achieved by
Euk-mPLoc 2.0 is more than 24% higher than those by any of the existing predictors. As a user-friendly
web-server, Euk-mPLoc 2.0 is freely accessible at
http://www.csbio.sjtu.edu.cn/bioinf/euk-multi-2/. For a query protein sequence of 400 amino acids, it will take about 15 seconds for
the web-server to yield the predicted result; the longer the sequence is, the more
time it may usually need. It is anticipated that the novel approach and the powerful
predictor as presented in this paper will have a significant impact to Molecular Cell
Biology, System Biology, Proteomics, Bioinformatics, and Drug Development.
Related collections
Most cited references59
- Record: found
- Abstract: found
- Article: not found
Gene Ontology: tool for the unification of biology
- Record: found
- Abstract: found
- Article: not found