- Record: found
- Abstract: found
- Article: found
iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins

Read this article at
Abstract
Predicting protein subcellular localization is an important and difficult problem,
particularly when query proteins may have the multiplex character, i.e., simultaneously
exist at, or move between, two or more different subcellular location sites. Most
of the existing protein subcellular location predictor can only be used to deal with
the single-location or “singleplex” proteins. Actually, multiple-location or “multiplex”
proteins should not be ignored because they usually posses some unique biological
functions worthy of our special notice. By introducing the “multi-labeled learning”
and “accumulation-layer scale”, a new predictor, called
iLoc-Euk, has been developed that can be used to deal with the systems containing both singleplex
and multiplex proteins. As a demonstration, the jackknife cross-validation was performed
with
iLoc-Euk on a benchmark dataset of eukaryotic proteins classified into the following 22 location
sites: (1) acrosome, (2) cell membrane, (3) cell wall, (4) centriole, (5) chloroplast,
(6) cyanelle, (7) cytoplasm, (8) cytoskeleton, (9) endoplasmic reticulum, (10) endosome,
(11) extracellular, (12) Golgi apparatus, (13) hydrogenosome, (14) lysosome, (15)
melanosome, (16) microsome (17) mitochondrion, (18) nucleus, (19) peroxisome, (20)
spindle pole body, (21) synapse, and (22) vacuole, where none of proteins included
has
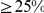 pairwise sequence identity to any other in a same subset. The overall success rate
thus obtained by
iLoc-Euk was 79%, which is significantly higher than that by any of the existing predictors
that also have the capacity to deal with such a complicated and stringent system.
As a user-friendly web-server,
iLoc-Euk is freely accessible to the public at the web-site
http://icpr.jci.edu.cn/bioinfo/iLoc-Euk. It is anticipated that
iLoc-Euk may become a useful bioinformatics tool for Molecular Cell Biology, Proteomics, System
Biology, and Drug Development Also, its novel approach will further stimulate the
development of predicting other protein attributes.
pairwise sequence identity to any other in a same subset. The overall success rate
thus obtained by
iLoc-Euk was 79%, which is significantly higher than that by any of the existing predictors
that also have the capacity to deal with such a complicated and stringent system.
As a user-friendly web-server,
iLoc-Euk is freely accessible to the public at the web-site
http://icpr.jci.edu.cn/bioinfo/iLoc-Euk. It is anticipated that
iLoc-Euk may become a useful bioinformatics tool for Molecular Cell Biology, Proteomics, System
Biology, and Drug Development Also, its novel approach will further stimulate the
development of predicting other protein attributes.
Related collections
Most cited references78
- Record: found
- Abstract: found
- Article: not found
Gene Ontology: tool for the unification of biology
- Record: found
- Abstract: found
- Article: not found
The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology.
- Record: found
- Abstract: found
- Article: not found