- Record: found
- Abstract: found
- Article: found
Excessive Cell Growth Causes Cytoplasm Dilution And Contributes to Senescence

Read this article at
Summary
Cell size varies greatly between cell types, yet within a specific cell type and growth condition, cell size is narrowly distributed. Why maintenance of a cell-type specific cell size is important remains poorly understood. Here we show that growing budding yeast and primary mammalian cells beyond a certain size impairs gene induction, cell-cycle progression, and cell signaling. These defects are due to the inability of large cells to scale nucleic acid and protein biosynthesis in accordance with cell volume increase, which effectively leads to cytoplasm dilution. We further show that loss of scaling beyond a certain critical size is due to DNA becoming limiting. Based on the observation that senescent cells are large and exhibit many of the phenotypes of large cells, we propose that the range of DNA:cytoplasm ratio that supports optimal cell function is limited and that ratios outside these bounds contribute to aging.
Graphical Abstract
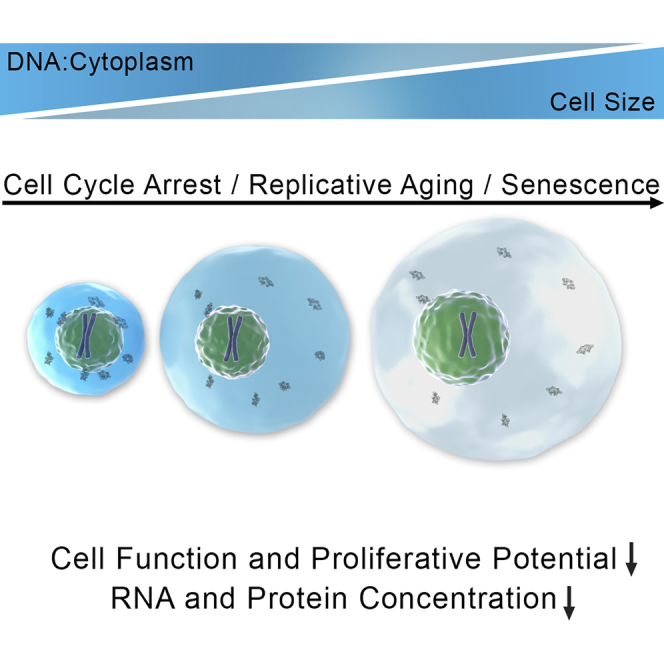
Highlights
-
•
Deviation from normal cell size interferes with cell function and proliferation
-
•
DNA becomes limiting for cell function if cells grow too large
-
•
Uncoupling of protein synthesis and volume causes cytoplasm dilution in big cells
-
•
Excessive cell growth contributes to functional decline in senescence
Abstract
Optimal cell function requires maintenance of a narrow range of DNA:cytoplasm ratios and when cell size exceeds this ratio cytoplasmic dilution contributes to senescence
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
Genomic expression programs in the response of yeast cells to environmental changes.
- Record: found
- Abstract: found
- Article: not found
MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes
- Record: found
- Abstract: found
- Article: not found