- Record: found
- Abstract: found
- Article: found
Schimke immunoosseous dysplasia: an ultra-rare disease. a 20-year case series from the tertiary hospital in the Czech Republic
Read this article at
Abstract
Background
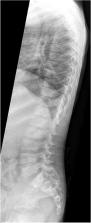
Schimke immunoosseous dysplasia (SIOD) is an ultra-rare inherited disease affecting many organ systems. Spondyloepiphyseal dysplasia, T-cell immunodeficiency and steroid resistant nephrotic syndrome are the main symptoms of this disease.
Case presentation
We aimed to characterize the clinical, pathological and genetic features of SIOD patients received at tertiary Pediatric Nephrology Center, University Hospital Motol, Prague, Czech Republic during the period 2001–2021. The mean age at diagnosis was 21 months (range 18–48 months). All patients presented with growth failure, nephropathy and immunodeficiency. Infections and neurologic complications were present in most of the affected children during the course of the disease.
Related collections
Most cited references24

- Record: found
- Abstract: found
- Article: found
The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies
- Record: found
- Abstract: not found
- Article: not found