- Record: found
- Abstract: found
- Article: found
Computation-aided studies related to the induction of specialized metabolite biosynthesis in microbial co-cultures: An introductory overview
Read this article at
Abstract
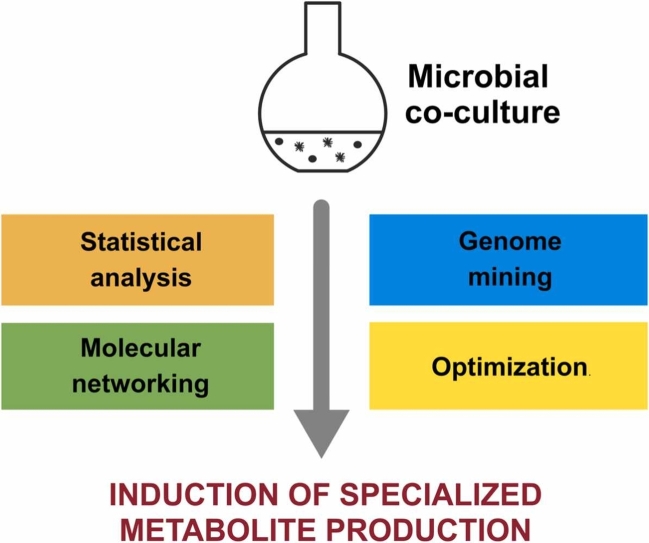
Co-cultivation is an effective method of inducing the production of specialized metabolites (SMs) in microbial strains. By mimicking the ecological interactions that take place in natural environment, this approach enables to trigger the biosynthesis of molecules which are not formed under monoculture conditions. Importantly, microbial co-cultivation may lead to the discovery of novel chemical entities of pharmaceutical interest. The experimental efforts aimed at the induction of SMs are greatly facilitated by computational techniques. The aim of this overview is to highlight the relevance of computational methods for the investigation of SM induction via microbial co-cultivation. The concepts related to the induction of SMs in microbial co-cultures are briefly introduced by addressing four areas associated with the SM induction workflows, namely the detection of SMs formed exclusively under co-culture conditions, the annotation of induced SMs, the identification of SM producer strains, and the optimization of fermentation conditions. The computational infrastructure associated with these areas, including the tools of multivariate data analysis, molecular networking, genome mining and mathematical optimization, is discussed in relation to the experimental results described in recent literature. The perspective on the future developments in the field, mainly in relation to the microbiome-related research, is also provided.
Graphical Abstract
Related collections
Most cited references121
- Record: found
- Abstract: found
- Article: not found
Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding
- Record: found
- Abstract: found
- Article: not found
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges
- Record: found
- Abstract: found
- Article: not found
Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking.
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.