- Record: found
- Abstract: found
- Article: found
Mitochondrial composition and function under the control of hypoxia
review-article
24 February 2017
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
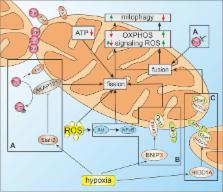
Hypoxia triggers several mechanisms to adapt cells to a low oxygen environment. Mitochondria are major consumers of oxygen and a potential source of reactive oxygen species (ROS). In response to hypoxia they exchange or modify distinct subunits of the respiratory chain and adjust their metabolism, especially lowering the citric acid cycle. Intermediates of the citric acid cycle participate in regulating hypoxia inducible factors (HIF), the key mediators of adaptation to hypoxia. Here we summarize how hypoxia conditions mitochondria with consequences for ROS-production and the HIF-pathway.
Graphical abstract

Highlights
Related collections
Most cited references67
- Record: found
- Abstract: found
- Article: not found
Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia.
Huafeng Zhang, Marta Bosch-Marce, Larissa Shimoda … (2008)
- Record: found
- Abstract: found
- Article: not found
Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases.
Hideaki Kamata, Shi-Ichi Honda, Shin Maeda … (2005)
- Record: found
- Abstract: found
- Article: not found
Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability.
David R Wise, Patrick Ward, Jessica Shay … (2011)