- Record: found
- Abstract: found
- Article: found
De novo sequencing of sunflower genome for SNP discovery using RAD (Restriction site Associated DNA) approach
Read this article at
Abstract
Background
Application of Single Nucleotide Polymorphism (SNP) marker technology as a tool in sunflower breeding programs offers enormous potential to improve sunflower genetics, and facilitate faster release of sunflower hybrids to the market place. Through a National Sunflower Association (NSA) funded initiative, we report on the process of SNP discovery through reductive genome sequencing and local assembly of six diverse sunflower inbred lines that represent oil as well as confection types.
Results
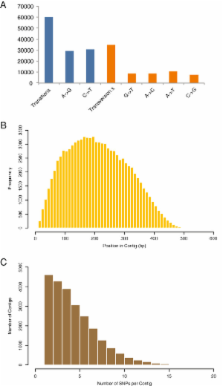
A combination of Restriction site Associated DNA Sequencing (RAD-Seq) protocols and Illumina paired-end sequencing chemistry generated high quality 89.4 M paired end reads from the six lines which represent 5.3 GB of the sequencing data. Raw reads from the sunflower line, RHA 464 were assembled de novo to serve as a framework reference genome. About 15.2 Mb of sunflower genome distributed over 42,267 contigs were obtained upon assembly of RHA 464 sequencing data, the contig lengths ranged from 200 to 950 bp with an N 50 length of 393 bp. SNP calling was performed by aligning sequencing data from the six sunflower lines to the assembled reference RHA 464. On average, 1 SNP was located every 143 bp of the sunflower genome sequence. Based on several filtering criteria, a final set of 16,467 putative sequence variants with characteristics favorable for Illumina Infinium Genotyping Technology (IGT) were mined from the sequence data generated across six diverse sunflower lines.
Related collections
Most cited references43
- Record: found
- Abstract: found
- Article: not found
The complete genome of an individual by massively parallel DNA sequencing.
- Record: found
- Abstract: found
- Article: not found
SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries.
- Record: found
- Abstract: found
- Article: not found