- Record: found
- Abstract: not found
- Article: not found
CRISPR-assisted detection of RNA–protein interactions in living cells
Author(s):
Wenkai Yi ,
Jingyu Li ,
Xiaoxuan Zhu ,
Xi Wang ,
Ligang Fan ,
Wenju Sun ,
Linbu Liao ,
Jilin Zhang ,
Xiaoyu Li ,
Jing Ye ,
Fulin Chen ,
Jussi Taipale ,
Kui Ming Chan ,
Liang Zhang ,
Jian Yan
Publication date (Electronic):
June 22 2020
Journal:
Nature Methods
Publisher:
Springer Science and Business Media LLC
Read this article at

There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Related collections
Most cited references17
- Record: found
- Abstract: found
- Article: not found
Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments.
Rainer Breitling, Patrick Armengaud, Anna Amtmann … (2004)
One of the main objectives in the analysis of microarray experiments is the identification of genes that are differentially expressed under two experimental conditions. This task is complicated by the noisiness of the data and the large number of genes that are examined simultaneously. Here, we present a novel technique for identifying differentially expressed genes that does not originate from a sophisticated statistical model but rather from an analysis of biological reasoning. The new technique, which is based on calculating rank products (RP) from replicate experiments, is fast and simple. At the same time, it provides a straightforward and statistically stringent way to determine the significance level for each gene and allows for the flexible control of the false-detection rate and familywise error rate in the multiple testing situation of a microarray experiment. We use the RP technique on three biological data sets and show that in each case it performs more reliably and consistently than the non-parametric t-test variant implemented in Tusher et al.'s significance analysis of microarrays (SAM). We also show that the RP results are reliable in highly noisy data. An analysis of the physiological function of the identified genes indicates that the RP approach is powerful for identifying biologically relevant expression changes. In addition, using RP can lead to a sharp reduction in the number of replicate experiments needed to obtain reproducible results.
- Record: found
- Abstract: found
- Article: not found
Suppression of progenitor differentiation requires the long noncoding RNA ANCR.
Markus Kretz, Dan Webster, Ross Flockhart … (2012)
Long noncoding RNAs (lncRNAs) regulate diverse processes, yet a potential role for lncRNAs in maintaining the undifferentiated state in somatic tissue progenitor cells remains uncharacterized. We used transcriptome sequencing and tiling arrays to compare lncRNA expression in epidermal progenitor populations versus differentiating cells. We identified ANCR (anti-differentiation ncRNA) as an 855-base-pair lncRNA down-regulated during differentiation. Depleting ANCR in progenitor-containing populations, without any other stimuli, led to rapid differentiation gene induction. In epidermis, ANCR loss abolished the normal exclusion of differentiation from the progenitor-containing compartment. The ANCR lncRNA is thus required to enforce the undifferentiated cell state within epidermis.
- Record: found
- Abstract: found
- Article: found
AlleleSeq: analysis of allele-specific expression and binding in a network framework
Joel Rozowsky, Alexej Abyzov, Jing-jing Wang … (2011)
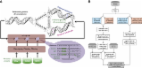
Introduction Due to rapidly increasing throughput and decreasing costs, next-generation short read sequencing is fast replacing array-based technology for performing functional genomic assays such as mapping locations of transcription factor binding or determining transcribed sequences in the genome. The initial analyses of high-throughput functional data using ChIP-Seq (Johnson et al, 2007; Robertson et al, 2007) or RNA-Seq (Mortazavi et al, 2008; Nagalakshmi et al, 2008) yield similar results that were obtained using tiling array-based methodologies albeit with greater sensitivity and resolution, that is, binding regions or regions of transcription. Also, with the developments in sequencing technologies there have been increasingly larger studies of the amount of sequence variation across the human population (The 1000 Genomes Project Consortium, 2010). A natural area of recent focus has been looking at the degree of functional genomic differences across the human population (Gregg et al, 2010a, 2010b; Kasowski et al, 2010; McDaniell et al, 2010; Montgomery et al, 2010; Pickrell et al, 2010). However, in order to understand population effects it is first useful to characterize the effects of functional variation within a single individual such as differences of expression and binding between alleles (i.e., allele-specific differences). When comparing functional data between individuals it is necessary to worry about normalization before any comparisons are performed; however, within a single individual there is a natural control of each allele against each other. By utilizing the actual sequence composition of the functional genomic sequence reads that overlap a heterozygous SNP, it is possible to determine the sequences that originate from each allele separately (Degner et al, 2009; McDaniell et al, 2010; Montgomery et al, 2010; Pickrell et al, 2010; Lalonde et al, 2011). Thus, it is possible to determine sites where transcription or transcription factor binding is originating predominately from one allele, that is, allele-specific expression (ASE) or allele-specific binding (ASB); however, there are number of technical issues which make this analysis challenging. In each of the recently published studies that contained some level of allele-specific analysis, only one type of functional genomic assay was performed. A logical question is how these allele-specific events are coupled between assays. At first glance, we expect significant coordination between binding of different transcription factors and expression of target genes. This has been previously been studied in a more limited manner using array-based technologies (Maynard et al, 2008). Here, we address this question by analyzing a number of different functional genomic data sets using a pipeline that we have developed, AlleleSeq, for determining sites showing allele-specific behavior. For the first time, we analyze allele-specific behavior for both transcription data using a very deeply sequenced RNA-Seq data set (∼160 million mapped reads) as well as matching deeply sequenced ChIP-Seq data sets (∼30 to ∼60 million mapped reads) for a number of different transcription factors (cFos, cMyc, JunD, Max, NfκB, and CTCF) as well as polymerases (RNA Polymerase II and Polymerase III). These experiments were generated for the lymphoblastoid cell line GM12878, which has also been deeply sequenced together with both parents (as a trio) as part of the pilot II phase of The 1000 Genomes Project Consortium (2010). Thus, for these data sets we have a complete set of heterozygous variants (SNPs, indels, and structural variations (SVs)) for the individual NA12878, which can mostly be phased into maternal and paternal variants by comparing against the parents sequences. This is important for assessing the genome-wide amount of allele-specific behavior, which is severely limited by the number of identified heterozygous SNPs available (for instance, Montgomery et al (2010) and Pickrell et al (2010) used HapMap III SNP calls which are ∼10-fold fewer than those available from pilot II of the 1000 Genomes Project). Allele-specific behavior is presumably occurring also in regions devoid of heterozygous SNPs, where we cannot distinguish between the alleles. When assessing the number of comparisons of allele-specific behavior between transcription factor binding and expression, 10-fold fewer total number of heterozygous SNPs would only allow for ∼100-fold fewer comparisons between ASB and ASE SNPs to be made. There are numerous technical hurdles in determining allele-specific behavior. One might think that it is possible to simply map the sequenced reads against the reference genome in order to determine allele differences; however, this introduces reference biases. Most analyses of human genomic data use the reference genome sequence for comparison; nevertheless, when genome scale analysis of allele-specific behavior is performed we show that it is necessary to align reads against a diploid sequence for that individual. We deal with this by constructing a diploid personal genome sequence by using the variation data (both for SNPs, indels, and SVs) for NA12878 (Mills et al, 2011; The 1000 Genomes Project Consortium, 2010). While the 1000 Genomes Project has created call sets of sequence variants for each of the different genomes sequenced, they have not however assembled genome sequences (including NA12878) for each of the individuals sequenced. In the first part of our AlleleSeq pipeline, we generate a diploid genome sequence of maternal and paternal haplotypes by integrating the phased variation data (SNPs, indels, and SVs) onto the reference genome sequence. In addition, we filter out genomic sequences that are likely to correspond to copy number variants (CNVs) using read-depth analysis (Abyzov et al, 2011). Construction of individual personal reference diploid sequences, as a first step for functional genomic analysis, will likely become standard in the near future. In this paper, we show that ASE of genes as well as novel transcribed regions, that is, novel transcriptionally active regions (TARs) or transfrags (Kapranov et al, 2002; Rinn et al, 2003; Bertone et al, 2004), are coordinated with ASB of transcription factors and other DNA binding proteins located adjacent to the transcribed region. One can measure how well ASB and ASE are coordinated, by using a correlation plot of the two. However, representing the coordination between multiple allele-specific events is difficult. In order to facilitate this, we show how ASB for multiple transcription factors is coordinated with ASE of the target genes or novel TARs by visualizing this behavior using a simplifying regulatory network. We will see how certain allele-consistent regulatory motifs are enriched using network analysis. We will observe that ASB and ASE are not as coordinated as might have been naively expected and speculate on potentially complexities of allele-specific regulation. Results We start by assembling a set of sequence variants from the 1000 Genomes Project for the NA12878 individual. We then generated deeply sequenced ChIP-Seq data sets for cFos, cMyc, JunD, Max, and RNA Polymerase II for the GM12878 cell line. We also created a matching deeply sequenced RNA-Seq data set for the same cell line. We combined these data sets with previously published matching data sets for RNA Polymerase II, RNA Polymerase III, NfκB, and CTCF (Kasowski et al, 2010; McDaniell et al, 2010; Raha et al, 2010). We summarize these data sets in Table I (see Materials and methods for further details). Determining allele-specific behavior from functional genomic data alone Intuitively if one has performed a deeply sequenced functional genomic experiment such as RNA-Seq or ChIP-Seq from a single individual, it should be possible to determine allele-specific behavior solely from the sequences obtained. The first step in this approach would be to determine the SNPs and other sequence variants directly from the sequence reads obtained. This might be true for certain regions sequenced at great depth; however, since functional genomic data (e.g., reads from a ChIP-Seq experiment) cover the genome with greatly varying sequence depth due to the nature of the functional assay. Thus, the accuracy of SNP (and other variant) calling from functional genomics data will necessarily vary across the genome. Conversely, the average sequencing depth across the genome for conventional genomic DNA sequencing is nearly uniform (with some differences to repeated regions and compositional biases). We find that the accuracy of de novo SNP calling using reads from a functional genomic sequencing experiment such as RNA-Seq using a standard SNP caller package (e.g., SNVmix; Shah et al, 2009) is not as good as we would need for determining allele-specific behavior (see Supplementary Table 1 for the results of de novo SNP calling of heterozygous SNPs). Any significant amount of miscalling of heterozygous SNPs will (obviously) lead to ill determined allele-specific behavior. There are a number of possible explanations for such miscalls; the very events we would like to find, SNPs within regions showing ASE could potentially appear as homozygous using only the RNA-Seq sequence reads. If one experimentally only obtains sequences from a region that is expressed on one allele (due to ASE) then there is no way to know that any base within that region is polymorphic. Second, RNA editing could also lead to variations in RNA sequences that are not present at the DNA level. Finally, sequencing of RNA involves additional experimental steps like usage of reverse transcriptase that can increase chance of mis-sequencing. Obviously, determining short indels from sequenced functional genomic data would be even harder and SVs would be nearly impossible. Thus, while it might be possible to determine certain sequence variants from the functional genomic sequence reads, in order to generate a comprehensive set of polymorphic sites as well as other forms of sequence variation it is necessary to have an independently determined set from sequenced genomic DNA (such as from the 1000 Genomes Consortium). Building an individual diploid reference genome for NA12878 It might not seem obvious but for a number of reasons reconstruction of a diploid personal genome sequence and using it instead of the reference genome is a critical step preceding allele-specific analysis. First, using reference genome introduces biases in read mapping—reads originated from non-reference allele are more susceptible to mismapping since, when aligned to the reference allele, they contain at least one mismatch (in case of SNPs) or gap (in case of indels)—the reference bias effect, that is, both alleles are not treated equally by default. Second, expression or binding in regions of genome SV could be misinterpreted as ASE or ASB. For example, duplication of an allele in the studied genome will double binding signal for the allele while signal for the allele on another haplotype will be unchanged. Last, but not least, SNP calling in the regions of SV is likely to be less precise and contain more false positives compared with non-SV regions (The 1000 Genomes Project Consortium, 2010). Thus, we construct a personal diploid genome of NA12878 (see Materials and methods), by utilizing genomic variations (see Table II for summary statistics) determined in the framework of The 1000 Genomes Project Consortium (2010) and, additionally, SVs determined by sequencing of fosmid clones (Kidd et al, 2008). To accomplish this, we have developed a tool—vcf2diploid—for personal genome construction, which constitutes the first part of the AlelleSeq pipeline (see Figure 1A). The tool uses as input VCF files with all the SNPs, indels, and SVs available for an individual of interest and outputs fasta sequences for each allele for each chromosome, along with equivalence map files (see Figure 1 and Supplementary Figure 1) that map nucleotide positions between paternal, maternal, and reference haplotypes. It is important to be able to map annotation (i.e., genes) from the reference genome to the personal genome sequences. This is done using chain files, which facilitate the mapping of annotated regions between genomes using the liftOver tool (Rhead et al, 2010). This is particularly important for RNA-Seq where we also build maternal and paternal versions of the gene annotation (including, most importantly, splice-junction library) by mapping the GENCODE annotation (GENCODE 3c annotation is available from the UCSC Genome Browser; Harrow et al, 2006) onto the personal diploid genome. The constructed diploid genome of NA12878 was different in 3 962 637 (∼0.14%) bases from the reference for paternal and in 3 947 162 (∼0.14%) for maternal alleles. The software package to perform personal genome sequence construction (the vcf2diploid tool and associated source code), the actual diploid sequence for NA12878, splice-junction sequences and personalized gene annotation for NA12878 and corresponding equivalence maps (between the maternal and paternal sequences as well as the reference genome, NCBI36/hg18) are available from http://alleleseq.gersteinlab.org. The diploid sequence for NA12878 is a valuable resource for anyone performing any sequence-based analysis on this genome. The GM12878 cell lines are a primary tier one cell line under detailed investigation by the ENCODE Consortium. It should be also noted that a constructed personal genome is only as good and as complete as the variants used in construction. In light of this, the diploid genome of NA12878 that is presented here, is not perfect, but we believe it is the best possible sequence to date since it includes the most comprehensive set of variants. We intend to update this assembly as a resource, as sequence variants are even more accurately determined. In order to assess the effect of the differences between the maternal and paternal sequences compared with using the reference genome sequence on functional genomic data, we aligned the reads from the Pol II and CTCF ChIP-Seq data for GM12878 against each of the three sequences using BOWTIE (Langmead et al, 2009; see Supplementary Figure 2). In Table III, we compare the Pol II reads that align to each of the three genome sequences (reference, maternal, and paternal haplotypes). We observe that by allowing up to two mismatches more reads (0.3% for paternal and 0.4% for maternal) align to the correct NA12878 as compared with the reference genome sequence (NCBI36). The major difference in numbers for paternal/maternal and reference haplotypes is due to reads that map to one haplotype but not the other. Namely, only about 0.1–0.2% of reads that map to the reference cannot be mapped to paternal or maternal haplotype, while a significantly higher fraction of reads (∼0.5%) map to the paternal or maternal genome and cannot be mapped to the reference. For paternal and maternal haplotypes, unmapped reads and reads with different mapping locations contribute roughly equally to the differences in overall mapping, presumably mostly due to short indels and SVs. We also see similar results for the same analysis done to the reads for CTCF ChIP-Seq (see Supplementary Table 2). This demonstrates that it is important to use a correctly assembled personal genome for aligning reads when performing an allele specificity analysis. Similarly, transcription factor binding sites also overlapped more when aligned to the maternal and paternal genomes of NA12878, rather than the reference sequence. For this comparison, we used the set of independently mapped reads for all three genome sequences to determine binding sites using PeakSeq (Rozowsky et al, 2009), and performed a pair-wise nucleotide overlap of the binding sites between the three genome sequences (Supplementary Table 3). In addition, we observe that the differences in binding sites, among the three genomes, are greater than the underlying differences in read mapping. Determining ASE and ASB The second part of the AlleleSeq pipeline determines ASE using RNA-Seq data and ASB using ChIP-Seq data. After the maternal- and paternal-derived haploid sequences are constructed, sequenced reads are aligned against the maternal and paternal sequences separately using BOWTIE (Langmead et al, 2009). Locations of mapping are determined by selecting the best alignment to both genome sequences. Read counts for the maternal and paternal alleles are then generated at each heterozygous SNP nucleotide positions, and ASE/ASB events are reported by applying a binomial test followed by correction for multiple hypothesis testing. SNP positions that by read-depth analysis (Abyzov et al, 2011) are determined to be potentially in a CNV (the read depth of genomic DNA reads in a 1-kb window around the SNP is either 3) are excluded (see Materials and methods). We correct for multiple hypothesis testing by estimating the false-discovery rate (FDR) by explicit simulation of the number of false positives given an even null background (i.e., no allele-specific events)—see Figure 1B for a schematic of the second part of the pipeline (see Materials and methods for further technical details). We also align reads to the maternal and paternal splice-junction libraries and determine splice-junction ASE SNPs in a similar way. Results for GM12878 RNA-Seq and ChIP-Seq data We start our study of allele-specific phenomena by first focusing on analyses of individual events that occur within single experimental data set. We then analyze the coordination between binding and expression in a pair-wise manner using direct correlation. Finally, we investigate higher order coordination of ASB and ASE using a regulatory network framework that will allow us to study the agreement between multiple regulatory interactions and target expression simultaneously. We summarize the results of the AlleleSeq pipeline applied to the RNA-Seq data and ChIP-Seq data for GM12878 in Table IV. In the upper half of the table, we present the results for all the autosomes and in the lower half for chromosome X (with all the transcription factor combined). In the second column of Table IV, we list the number of genomic elements (genes, novel TARs, and binding sites) that are accessible for allele-specific behavior—that is, those that we could have detected allele-specific activity in. This is the set of genomic elements that contain at least one heterozygous SNP and are sequenced at sufficient depth in order to detect allele-specific activity, see Materials and methods for further details. The third column shows that number of genomic elements that we determine to show allele-specific behavior. The fourth column shows the fraction of genomic elements that are accessible for allele-specific behavior that do show either ASE or ASB. Finally for allele-specific events that can be phased we report those that are maternal or paternal. We observe that ∼19.4% of all autosomal GENCODE genes that are accessible for allele-specific behavior exhibit ASE. We similarly find that 21.6% of accessible heterozygous SNPs within splice junctions of genes also show ASE. Similarly, we find that 9.3% of autosomally expressed accessible novel TARs show ASE, we expect this number to be lower than genes as novel TARs correspond to exons of genes. We found that for genes that contained two or more heterozygous SNP showing allele-specific behavior, >81% of the time all the SNPs would show consistent ASE from the same allele (significantly greater than expected by chance), some of the exceptions could be due to allele-specific alternate splicing. For the transcription factors, the fraction of accessible autosomal binding sites that exhibit allele-specific behavior varies between 2% (for cMyc) and 11% (for Pol II). A possible explanation for the difference between binding and expression allele specificity is that even though these ChIP-Seq data sets have been sequence quite deeply (see Table I), in order to have comparable power with the RNA-Seq data one would need to sequence an order or magnitude or two further. The number of overlapping sequence reads in binding site peaks for ChIP-Seq data depends on the efficiency of the antibody used and for most ChIP-Seq data sets we do not have sufficient read depth within a binding site as compared with the read depth within exons of highly expressed genes. As expected when restricted to the autosomes, both ASE for genes and novel TARs and ASB for all the transcription factors and polymerases are evenly divided between the maternal and paternal alleles. In the lower half of Table IV, we present similar results for chromosome X. Since NA12878 is female there are two copies of chromosome X. We first observe that almost 80% of the accessible genes on chromosome X exhibit allele-specific behavior and 95% of these are expressed on the maternal copy. This is consistent with our knowledge of X-chromosome inactivation (Lyon, 1961; Goto and Monk, 1998) where one copy of the two copies is shut off. There are four genes on chromosome X that show ASE on the paternal copy; however, all of these are located in the pseudo-autosomal region of chromosome X which is known to escape X-chromosome inactivation (these include Xist, SLC25A6 and SFRS17A). We observe similar results on chromosome X for the allele-specific behavior of novel TARs as well as transcription factor binding where a greater fraction of sites exhibit allele-specific behavior compared with the autosomes and almost all are expressed on the maternal copy. It is interesting to note that most of the novel transcription and binding that shows paternal allele specificity are also in the pseudo-autosomal region similar to Xist and possibly have an associated functional role. We make available the complete list of SNPs that show ASB or ASE in VCF format as a resource from our website http://alleleseq.gersteinlab.org. We imagine that this file may be a useful resource for researchers interested in focusing on allele-specific sites in less deeply sequenced functional genomic experimental data sets. This might be valuable even if the functional genomic experiment was not performed on the GM12878 cell line as regions to investigate for allele-specific behavior. There are a number of technical issues associated with determining allele-specific behavior including various biases that can be introduced as part of the analysis. We investigate some of these potential effects in detail below: Reference bias: In order to assess whether our pipeline has some residual bias toward the reference allele versus the alternate, we can plot the fraction of reads from the alternative allele for each heterozygous SNP location sequenced sufficiently deeply. If there were no bias, we would expect that this distribution would be symmetric having as many reference allele-specific locations as for the alternate allele. In Figure 2, we plot the alternative allele fraction distribution for the RNA-Seq data, Pol II, and the other transcription factors combined. We first observe that the overall distributions are fairly symmetric and that the allele-specific events (indicated in blue) are also symmetric—this indicates that there is no residual bias toward or against the reference allele. In Supplementary Figure 3, we observe similar distributions for the fractions of reads from the maternal allele for each heterozygous SNP location that could be phased and that was sequenced sufficiently deeply in the appropriate assay. Correlation with SNP quality (genotype likelihood scores): SNPs determined by The 1000 Genomes Project Consortium (2010) each have a genotype likelihood score. In Supplementary Figure 4, we plot the distribution of all heterozygous SNPs and the subset of ASE SNPs versus this genotype likelihood score. We see a slight enrichment of ASE SNPs will lower scores; however, the majority of SNPs from both distributions have the highest possible score. For comparison, non-synonymous SNPs also show a similar distribution. Relation to genome duplications (effect of Degner et al, 2009): It has been reported by Degner et al (2009) that heterozygous sites showing apparent allele-specific behavior can be caused by regions in the genome that have been duplicated. Thus, even though there might be a similar number of reads coming from each allele, only one of the alleles would have uniquely mapping reads which would lead to seemingly allele-specific behavior (see Supplementary Figure 5 for a schematic comparing region without a duplication to regions that have been duplicated). In order to assess the size of this effect on our results, we used the Pol II ChIP-Seq reads mapped uniquely and independently to each of the maternal and paternal genomes. This is as opposed to the normal mapping procedure in the AlleleSeq pipeline, where we map independently to both haplotypes and then select the allele with the better mapping location. At each heterozygous SNP location determined to show ASB we computed the haplotype fraction, the fraction of reads mapped to one allele over the sum of reads mapped to both alleles (we choose the haplotype fraction corresponding to the allele that has the greater fraction, see Supplementary Figure 5). For sites that have not been duplicated the haplotype fraction should have a value close to 0.5, while for duplicated regions exhibiting the Degner effect the fraction would be close to 1 (where all the uniquely mapped reads would come from one allele). In Supplementary Figure 6, we plot the distribution of haplotype fractions for all Pol II ASB sites. We observe that only a minority of the sites ( 0.6). As valid ASB sites that contain additional proximal sequence variants (such as additional SNPs or indels) would also exhibit a fraction biased toward one haplotype, we conclude that this is an upper bound on the size of this effect and do not consider it significant. Modified binomial test: In order to assess the effect of aligning reads against the constructed diploid genome sequence for NA12878 versus using the reference genome sequence we perform the following analysis. For the RNA-Seq reads, we also aligned the reads against the reference genome and determined ASE using an even binomial distribution (threshold applied in a similar manner). As an additional comparison, we also applied the methodology of Montgomery et al (2010) where a skewed binomial distribution is used with the reads aligned against the reference genome (they composite for the reference bias induced by mapping to the reference genome by modifying the binomial distribution). Similar to Figure 2, we plotted the distribution of all expressed heterozygous SNPs (ASE SNPs in blue) for these two methods in Supplementary Figure 7. Using the naive methodology with an even binomial we see the skew of the ASE SNPs toward the reference genome which is largely removed using the modified binomial test. When comparing our set of 5862 ASE SNPs determined using the personal genome we find that only 69% are shared with those determined using the naive approach. Using the modified binomial methodology from Montgomery et al (2010), we see an improvement (75% in common); however, we still are detecting a significant number of ASE sites that were missed aligning to the reference genome and only modifying the binomial test versus using the correct diploid genome to align against. Comparison with McDaniell et al, (2010): We have also compared the ASB sites for the CTCF ChIP-Seq data from the AlleleSeq pipeline against those from McDaniell et al (2010). Restricting the comparison with common heterozygous SNP between the two analysis (McDaniell et al, 2010 used an earlier set of SNP calls for NA12878 from The 1000 Genomes Project Consortium, 2010) we find that greater using a P-value threshold of 0.01 on their results >85% of the ASB SNPs are in common. Allele-specific elements using heterozygous indels: The AlleleSeq pipeline determines allele-specific behavior for genomic elements (transcribed regions or binding sites) that contain heterozygous SNPs. It is also possible to determine allele-specific behavior for genomic elements that contain a heterozygous indel. In Supplementary Table 4, we show the results for transcribed exons and novel TARs as well binding sites for Pol II and CTCF that can be determined to show allele-specific behavior using a heterozygous indel to distinguish the haplotypes. Correlation of ASB in binding sites containing known motifs In our analysis of ASB events, heterozygous SNPs are initially only used for distinguishing between the maternal and paternal alleles (presumably allele-specific behavior also occurs in genomic regions not containing heterozygous SNPs). However, in some locations the heterozygous SNP might be the causative reason for the difference in binding between the alleles, this would most likely occur in ASB sites where the heterozygous SNP is within a known DNA binding motif for a transcription factor. In order to see how ASB is correlated with perturbations to known DNA binding motifs, we compared the allele that is bound against the allele that matches better with the known motif. Thus, we first scanned ASB sites for known motifs, position weight matrices (PWMs) obtained from TRANSFAC (Matys et al, 2006) and JASPAR (Portales-Casamar et al, 2010) (see Materials and methods for further details). We correlated the nucleotide of the allele, which is preferentially bound with the fitness score of the PWM. We observed a number of significant correlations between the PWM score for both alleles and the allele that is bound. The allele that is bound tends to correspond to the better match to the known PWM. In particular, we see this for the known cMyc motif within both cMyc and Max binding sites (see Figure 3). This is interesting as we observe a correlation between the allele-specific behavior of cMyc motifs with Max binding sites, indicating that these transcription factors tend to significantly regulate the same target genes. We also include in Supplementary Figure 8 additional examples of the correlation between motif fitness score and the allele being bound for CTCF binding sites containing CTCF motifs and cMyc motifs within Pol II binding sites. Correlation of ASE with protein structural fitness Some heterozygous SNPs within genes can result in one allele losing its ability to function as a transcript (i.e., heterozygous loss of function). Additionally, non-synonymous SNPs within the protein-coding sequence can cause a difference in the structure fitness of the protein coded from each allele. We studied the coordination between these effects and the genes that show ASE. We first investigated the overlap between genes that exhibit ASE with genes that show loss of function on one allele due to a premature stop codon, a frame-shift or a disruption of an intron–exon splice site (all caused by heterozygous SNPs). We find four cases of genes that show ASE as well as heterozygous loss of function and in all four cases the allele that is expressed is opposite to the allele that has lost its ability to code for a protein. We speculate that in some of these cases the transcript from the allele suffering from a disruption might be degraded due to non-sense-mediated decay, which, in turn, might cause the ASE from the other allele. Since some heterozygous SNPs that show ASE are within the protein-coding sequence of genes, it is natural to ask whether the allele that is expressed could track with allele-dependent structural changes (for SNPs in non-synonymous positions in the protein-coding sequence). However, it is not clear that we expect to find a correlation between structural fitness and ASE, as many of these SNPs are not selected for in the human population in any case. In order to assess whether the allele that is expressed (for genes showing ASE) is correlated with the allele containing mutations deleterious to protein structure we performed the following analysis. We compared the occurrences of ASE SNPs within genes where the SNP corresponds to a non-synonymous substitution within the protein-coding sequence of the gene. Using the tool SIFT (Ng and Henikoff, 2003), we can compare the preference of the allele that is expressed with the allele whose amino-acid sequence shows better structural fitness. We find that 20 of the 37 genes that meet these criteria show expression on the allele that has the protein sequence that has better fitness. While we find slightly more genes where the allele with better structural fitness occurs on the same allele that is expressed, this result is not significant. Correlating ASB with ASE In the upper panel of Figure 4, we present an example of the gene SKA3 on chromosome 13 which has multiple heterozygous SNPs within exons which show consistent maternal ASE which agrees with the maternal ASB of another heterozygous SNP within a Pol II binding site proximal to the 5′ end of the gene. In this example, we see coordinated maternal binding of Pol II with expression of the gene. In the lower panel of Figure 4, we present a similar example of a novel transcribed region on chromosome 4 where we see coordinated paternal binding of Pol II with the paternal expression of the novel TAR. These two examples show how ASB is coordinated with ASE for a known gene and a novel TAR. To investigate this trend, we assess to what extent allele-specific behaviors detected using heterozygous SNPs are coordinated on a genome-wide scale. In Table V, we tabulate the number of genes and novel TARs that have evidence for ASE and for proximal ASB. We also tabulate the total counts of genes that have a proximal binding site where both the gene and binding are jointly accessible for detecting allele-specific behavior. We perform a similar calculation for novel TARs and their proximal sites. In Table V, we present the tabulated counts for Pol II & Pol III and CTCF separately from all the other transcription factors combined. We find a number of genes (74 genes proximal to Pol II & Pol III sites, 29 genes proximal to CTCF binding sites, and 44 genes with proximal transcription factor sites other than CTCF) and novel TARs (55, 8, and 15 for Pol II & Pol III, CTCF, and remaining transcription factors, respectively) in which we see both ASB proximal to ASE. We separate CTCF from the remaining transcription factors because of its function as an insulator. While these numbers might seem relatively small, they reflect the low chance of having both a proximal binding site as well as an expressed gene with both of them jointly accessible for the detection of allele-specific activity. This underscores the need to sequence deeply and use a comprehensively determined set of SNPs or else we would have significantly fewer genes to be able to compare ASB and ASE. In order to assess the degree of coordinated allele-specific behavior for the 74 genes that exhibit ASE that have a proximal ASB Pol II binding site we performed the following analysis. For each gene, we tabulated the allele-specific behavior of the most significant ASE SNP versus the most significant ASB SNP (if there are more than one significant heterozygous allele-specific SNP present). In Table VI , we tabulate maternal and paternal ASB of binding sites against maternal and paternal ASE of proximal genes (we do this separately for Pol II & Pol III, CTCF, and the remaining transcription factors combined). We see that there is a statistically significant coordination between ASB of Pol II & Pol III to genes that exhibit ASE (Fisher's exact test, P-value=1.4e−3). Similarly, as seen in Table VI there is also a statistically significant correlation between the 45 genes that exhibit ASE with ASB for all the combined transcription factors excluding CTCF (Fisher's exact test, P-value=1.8e−5). We do not however, see a significant correlation of ASB with ASE for CTCF which is probably due to its role as an insulator. Further coordination of allele-specific behavior As a further way to assess the coordination of allele-specific events within genes, we combined all the ASE and ASB SNPs that occurred within a gene (from 2.5 kb upstream of the transcription start site (TSS) to the transcription termination site (TTS) including introns). Using only genes that contained >10 SNPs showing ASE or ASB we could compute the fraction of SNPs that show maternal specificity. Ideally, if all SNPs were perfectly coordinated then the fraction would be either zero or one. In the first panel of Figure 5, we see the actual distribution, most genes do show a high degree of coordination. Under a random null (where each ASE or ASB event could be maternal or paternal with equal chance) for the same genes each with the same number of SNPs, we would expect a null distribution of maternal fraction computational simulated in panel two of Figure 5. Using a Kolmolgorov–Smirnov test, we find significantly more coordination of ASB and ASE SNPs in genes than compared with the random null expectation (P-value=8.45e−8; see the third panel of Figure 5). Representing ASE and ASB behavior on a regulatory network In the previous analysis, we showed that binding and expression were correlated in a pair-wise manner. Next, we would like to investigate the coordination of allele-specific behavior between multiple factors and expression simultaneously. This is hard to represent in a two-dimensional correlation plot; thus, we have developed a simplified representation of ASE and ASB in a regulatory network framework. Looking at the occurrences of network motifs (Milo et al, 2002) in this framework allows us to measure the coordination of allele-specific behavior between multiple transcription factors and target expression. The network shown in Figure 6 represents the regulatory network of six transcription factors (cMyc, Max, cFos, JunD, NfκB, and CTCF) and two polymerases (Pol II and Pol III). The network discretizes the ASB events into allele-specific regulation of target genes and novel TARs and their ASE. The edges in the network represent ASB of the TF or polymerase to the target gene or novel TAR (red edges indicate predominantly maternal regulation, blue edges indicate paternal regulation, and gray edges indicate allele-specific regulation that could not be phased). ASE of the target genes is indicated by the color of the target gene or novel TAR (red—maternal, blue—paternal, and gray—unphased allele-specific behavior). Thus, the network contains all information on the allele specificity of the regulation and the expression of the targets. One can observe that there is a clear agreement between the allele specificity of the regulation and the expression of the target. When we tabulate the maternal/paternal regulation edges with maternal/paternal expressed genes or novel TARs (see first part of Table VII) we find that they are highly coordinated (P-value 3 (the normalization factor of 2 indicates the diploid nature of the human genome). We filtered out SNPs that are more likely to be false positives or may represent duplicated or deleted regions which would complicate calling allele-specific behavior. The allele-specific SNP processing pipeline The pipeline has four main inputs: one or more collections of unmapped reads, a set of SNP positions, a personal genome, a set of known genes, listing transcription starts and stops, and exon coordinates. The processing follows these steps. For each logical set of reads: (1) The reads are trimmed, if necessary, to remove ends that contain large numbers of errors and filtered to remove any reads containing N's. (2) SNP locations are converted to a standardized format that describes the alleles for all heterozygous SNPs in GM12878, including parental phasing, if possible. Phasing is possible for all heterozygous GM12878 SNPs except those in which both parents are also heterozygous. (3) The filtered reads are mapped, using bowtie, to the maternal and paternal genomes. Bowtie was invoked with these flags: --best --strata -v 2 -m 1, which returns only unique hits within a minimum number of mismatches, up to two. (4) The two sets of mapped reads are merged into a single set, with each read represented at most once, using the better mapping from the maternal or paternal haplotypes. If the two mappings for the same read tie in quality, one is chosen at random. (5) Using Het-SNP file and the mapped reads, allele counts are generated for each Het-SNP location. The resulting counts file contains the number of As, Cs, Gs, and Ts found in reads mapped over each SNP location. Various other values are also generated for each Het-SNP location, including reference allele, maternal/paternal allele (if determinable), major and minor allele, and a binomial P-value assuming a 50/50 probability of sampling each of two alleles. (6) In order to calculate the FDR, we perform an explicit computational simulation to correct for multiple hypothesis testing. We start with all the heterozygous SNP locations; for each SNP location, we randomly assign each mapped read in the data set to either allele. At a given P-value threshold (using the binomial test), we can determine the number of false positive allele-specific event calls (from the simulated data); and thus, we can determine the FDR as the number of false positive over the total number of observed positives. We require a FDR of 50% and at least twice as high as the second most frequent nucleotide. A double-degenerate code is used if the combined frequencies of two nucleotides are >75% but each of them is present in 0.01) and P(n) is the background frequency of allele n. In Figure 3, we plot this difference in motif scores, delta (maternal–paternal) against the fraction of maternally derived reads overlapping the same heterozygous SNP in the ASB site. In small number of cases where there are multiple SNPs in the TF motif region, the best one is chosen where if the maternal read count fraction greater than half the best is equal to the biggest delta, while if fraction is less than half the smallest delta is chosen. Building an allele-dependent regulatory network We decided to integrate the expression data for genes and TARs from the RNA-Seq experiment with the TF binding data from the (cFos, cMyc, JunD, Max, NfkB, CTCF, Pol II, and III) ChIP-Seq experiments into a regulatory network. In order to construct a regulatory network to determine the edge between a TF and a gene by assigning an ASB event to a target ASE gene if it lies within 2.5 kb upstream of the annotated TSS and the TTS. For ASE novel TARs we do not know which strand is being expressed, thus we associate ASB events that occur within 2.5 kb of either end of the novel TAR. If it is allele specific then it could be further classified into paternal, maternal, and unphased. The ‘unphased' category represents the case where the experiments show allele specificity but it cannot be phased. After constructing the edges between the TFs and gene/novel TARs in the network, we overlaid the gene/novel TAR ASE information onto the nodes. Each gene/novel TAR was categorized into three categories: paternal, maternal, or unphased ASE. After constructing the network, we performed a network motif analysis on it, the results of which are shown in Table VII. We analyzed the occurrences of MIMs where two TFs regulate the same gene/novel TAR and SIMs where a single TF regulates two different gene/novel TARs, taking into account the allele specificity of the regulation and the expression of the targets. Counting of occurrences of MIMs and SIMs was performed using Cytoscape (Cline et al, 2007). Supplementary Material Supplementary Material Supplementary Figures S1–8 and Supplementary Tables S1–4 Supplementary Dataset 1 Supplementary data files of allele-specific SNPs in vcf format. Supplementary Dataset 2 Supplementary data files of genes showing alelle-specific expression. Review Process File
Author and article information
Journal
Title:
Nature Methods
Abbreviated Title:
Nat Methods
Publisher:
Springer Science and Business Media LLC
ISSN
(Print):
1548-7091
ISSN
(Electronic):
1548-7105
Publication date
(Electronic):
June
22 2020
Article
DOI: 10.1038/s41592-020-0866-0
PubMed ID: 32572232
SO-VID: 46274d49-d3a6-4637-88be-92edec2d974b
Copyright © ©
2020
History
Data availability: