- Record: found
- Abstract: found
- Article: found
Questions and controversies: the role of necroptosis in liver disease
Read this article at
Abstract
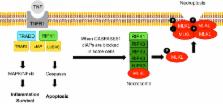
Acute and chronic liver injury results in hepatocyte death and turnover. If injury becomes chronic, the continuous cell death and turnover leads to chronic inflammation, fibrosis and ultimately cirrhosis and hepatocellular carcinoma. Controlling liver cell death both in acute injury, to rescue the liver from acute liver failure, and in chronic injury, to curb secondary inflammation and fibrosis, is of paramount importance as a therapeutic strategy. Both apoptosis and necrosis occur in the liver, but the occurrence of necroptosis in the liver and its contribution to liver disease is controversial. Necroptosis is a form of regulated necrosis which occurs in certain cell types when caspases (+/−cIAPs) are inhibited through the RIPK1-RIPK3 activation of MLKL. The occurrence of necroptosis in the liver has recently been examined in multiple liver injury models with conflicting results. The aim of this review is to summarize the published data with an emphasis on the controversies and remaining questions in the field.
Related collections
Most cited references73
- Record: found
- Abstract: found
- Article: not found
Mouse model of chronic and binge ethanol feeding (the NIAAA model).

- Record: found
- Abstract: found
- Article: found
Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death
- Record: found
- Abstract: found
- Article: not found