- Record: found
- Abstract: found
- Article: found
Reactive metabolite production is a targetable liability of glycolytic metabolism in lung cancer
Read this article at
Abstract
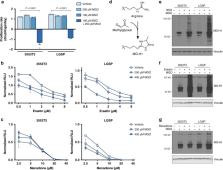
Increased glucose uptake and metabolism is a prominent phenotype of most cancers, but efforts to clinically target this metabolic alteration have been challenging. Here, we present evidence that lactoylglutathione (LGSH), a byproduct of methylglyoxal detoxification, is elevated in both human and murine non-small cell lung cancers (NSCLC). Methylglyoxal is a reactive metabolite byproduct of glycolysis that reacts non-enzymatically with nucleophiles in cells, including basic amino acids, and reduces cellular fitness. Detoxification of methylglyoxal requires reduced glutathione (GSH), which accumulates to high levels in NSCLC relative to normal lung. Ablation of the methylglyoxal detoxification enzyme glyoxalase I (Glo1) potentiates methylglyoxal sensitivity and reduces tumor growth in mice, arguing that targeting pathways involved in detoxification of reactive metabolites is an approach to exploit the consequences of increased glucose metabolism in cancer.
Abstract
Glycolysis is elevated in many cancers. In this study, the authors show that lactoylglutathione, a by-product of methylglyoxal produced from increased glycolysis, is elevated in lung cancer in mouse models and humans, arguing reactive metabolite production can be a liability for cancers.
Related collections
Most cited references38
- Record: found
- Abstract: found
- Article: not found
Lactate Metabolism in Human Lung Tumors
- Record: found
- Abstract: found
- Article: not found
An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma.
- Record: found
- Abstract: found
- Article: not found