- Record: found
- Abstract: found
- Article: found
Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer
Read this article at
Abstract
Background
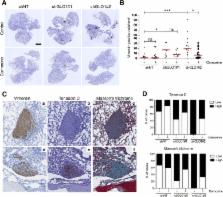
Elevated aerobic glycolysis rate is a biochemical alteration associated with malignant transformation and cancer progression. This metabolic shift unavoidably generates methylglyoxal (MG), a potent inducer of dicarbonyl stress through the formation of advanced glycation end products (AGEs). We have previously shown that the silencing of glyoxalase 1 (GLO1), the main MG detoxifying enzyme, generates endogenous dicarbonyl stress resulting in enhanced growth and metastasis in vivo. However, the molecular mechanisms through which MG stress promotes metastasis development remain to be unveiled.
Methods
In this study, we used RNA sequencing analysis to investigate gene-expression profiling of GLO1-depleted breast cancer cells and we validated the regulated expression of selected genes of interest by RT-qPCR. Using in vitro and in vivo assays, we demonstrated the acquisition of a pro-metastatic phenotype related to dicarbonyl stress in MDA-MB-231, MDA-MB-468 and MCF7 breast cancer cellular models. Hyperactivation of MEK/ERK/SMAD1 pathway was evidenced using western blotting upon endogenous MG stress and exogenous MG treatment conditions. MEK and SMAD1 regulation of MG pro-metastatic signature genes in breast cancer cells was demonstrated by RT-qPCR.
Results
High-throughput transcriptome profiling of GLO1-depleted breast cancer cells highlighted a pro-metastatic signature that establishes novel connections between MG dicarbonyl stress, extracellular matrix (ECM) remodeling by neoplastic cells and enhanced cell migration. Mechanistically, we showed that these metastasis-related processes are functionally linked to MEK/ERK/SMAD1 cascade activation in breast cancer cells. We showed that sustained MEK/ERK activation in GLO1-depleted cells notably occurred through the down-regulation of the expression of dual specificity phosphatases in MG-stressed breast cancer cells. The use of carnosine and aminoguanidine, two potent MG scavengers, reversed MG stress effects in in vitro and in vivo experimental settings.
Conclusions
These results uncover for the first time the key role of MG dicarbonyl stress in the induction of ECM remodeling and the activation of migratory signaling pathways, both in favor of enhanced metastatic dissemination of breast cancer cells. Importantly, the efficient inhibition of mitogen-activated protein kinase (MAPK) signaling using MG scavengers further emphasizes the need to investigate their therapeutic potential across different malignancies.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases.
- Record: found
- Abstract: found
- Article: not found
A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras.
- Record: found
- Abstract: found
- Article: not found