- Record: found
- Abstract: found
- Article: found
Lysosomal Ceramide Metabolism Disorders: Implications in Parkinson’s Disease
Read this article at
Abstract
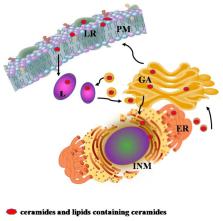
Ceramides are a family of bioactive lipids belonging to the class of sphingolipids. Sphingolipidoses are a group of inherited genetic diseases characterized by the unmetabolized sphingolipids and the consequent reduction of ceramide pool in lysosomes. Sphingolipidoses include several disorders as Sandhoff disease, Fabry disease, Gaucher disease, metachromatic leukodystrophy, Krabbe disease, Niemann Pick disease, Farber disease, and GM2 gangliosidosis. In sphingolipidosis, lysosomal lipid storage occurs in both the central nervous system and visceral tissues, and central nervous system pathology is a common hallmark for all of them. Parkinson’s disease, the most common neurodegenerative movement disorder, is characterized by the accumulation and aggregation of misfolded α-synuclein that seem associated to some lysosomal disorders, in particular Gaucher disease. This review provides evidence into the role of ceramide metabolism in the pathophysiology of lysosomes, highlighting the more recent findings on its involvement in Parkinson’s disease.
Related collections
Most cited references112
- Record: found
- Abstract: found
- Article: not found
Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson's disease.

- Record: found
- Abstract: found
- Article: found
Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations
- Record: found
- Abstract: found
- Article: not found