- Record: found
- Abstract: found
- Article: found
Current models of care for disorders of sex development – results from an International survey of specialist centres
Read this article at
Abstract
Background
To explore the current models of practice in centres delivering specialist care for children with disorders of sex development (DSD), an international survey of 124 clinicians, identified through DSDnet and the I-DSD Registry, was performed in the last quarter of 2014.
Results
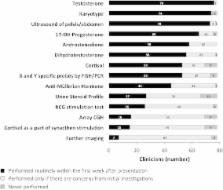
A total of 78 (63 %) clinicians, in 75 centres, from 38 countries responded to the survey. A formal national network for managing DSD was reported to exist in 12 (32 %) countries. The paediatric specialists routinely involved in the initial evaluation of a newborn included: endocrinologist (99 %), surgeon/urologist (95 %), radiologist (93 %), neonatologist (91 %), clinical geneticist (81 %) and clinical psychologist (69 %). A team consisting of paediatric specialists in endocrinology, surgery/urology, clinical psychology, and nursing was only possible in 31 (41 %) centres. Of the 75 centres, 26 (35 %) kept only a local DSD registry and 40 (53 %) shared their data in a multicentre DSD registry. Attendance in local, national and international DSD-related educational programs was reported by 69, 78 and 84 % clinicians, respectively. Participation in audits/quality improvement exercises in DSD care was reported by 14 (19 %) centres. In addition to complex biochemistry and molecular genetic investigations, 40 clinicians (51 %) also had access to next generation sequencing. A genetic test was reported to be more preferable than biochemical tests for diagnosing 5-alpha reductase deficiency and 17-beta hydroxysteroid dehydrogenase 3 deficiency by 50 and 55 % clinicians, respectively.
Related collections
Most cited references23
- Record: found
- Abstract: not found
- Article: not found
Consensus statement on management of intersex disorders.
- Record: found
- Abstract: found
- Article: found
Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care
- Record: found
- Abstract: found
- Article: not found