- Record: found
- Abstract: found
- Article: not found
The role of HYAL2 in LSS-induced glycocalyx impairment and the PKA-mediated decrease in eNOS–Ser-633 phosphorylation and nitric oxide production
Read this article at

Abstract
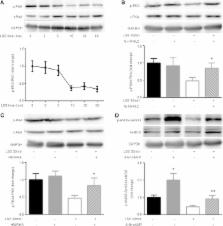
This study examines the mechanism of glycocalyx injury under low shear stress (LSS). LSS-induced changes of eNOS phosphorylation and NO production are based on the deficiency of the glycocalyx barrier.
Abstract
Hyaluronan (HA) in the endothelial glycocalyx serves as a mechanotransducer for high-shear-stress–stimulated endothelial nitric oxide synthase (eNOS) phosphorylation and nitric oxide (NO) production. Low shear stress (LSS) has been shown to contribute to endothelial inflammation and atherosclerosis by impairing the barrier and mechanotransduction properties of the glycocalyx. Here we focus on the possible role of hyaluronidase 2 (HYAL2) in LSS-induced glycocalyx impairment and the resulting alterations in eNOS phosphorylation and NO production in human umbilical vein endothelial cells (HUVECs). We show that LSS strongly activates HYAL2 to degrade HA in the glycocalyx. The dephosphorylation of eNOS–Ser-633 under LSS was triggered after HA degradation by hyaluronidase and prevented by repairing the glycocalyx with high–molecular weight hyaluronan. Knocking down HYAL2 in HUVECs protected against HA degradation in the glycocalyx by inhibiting the expression and activity of HYAL2 and further blocked the dephosphorylation of eNOS–Ser-633 and the decrease in NO production in response to LSS. The LSS-induced dephosphorylation of PKA was completely abrogated in HYAL2 siRNA–transfected HUVECs. The LSS-induced dephosphorylation of eNOS–Ser-633 was also reversed by the PKA activator 8-Br-cAMP. We thus suggest that LSS inhibits eNOS–Ser-633 phosphorylation and, at least partially, NO production by activating HYAL2 to degrade HA in the glycocalyx.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
The endothelial glycocalyx: composition, functions, and visualization
- Record: found
- Abstract: found
- Article: not found
Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface.
- Record: found
- Abstract: found
- Article: not found